Long QT Syndrome (LQTS)
Indice dell'articolo
What is Long QT Syndrome?
Long QT Syndrome (LQTS) is a genetically based cardiac pathology characterized by a high risk of ventricular cardiac arrhythmias, which can cause syncope (loss of consciousness), cardiac arrest, and sudden death. The disease is characterized by the prolongation of the QT interval, measured on the surface electrocardiogram, and by morphological anomalies of the T wave. LQTS manifests usually during childhood, but in some cases it may begin even after puberty. The severity of the disease is highly variable and, at least in part, dependent on the type of gene or mutation involved.
LQTS was first recognized in 1957 in Norway by Jervell and Lange-Nielsen, who described the subtype of the syndrome associated with neurological deafness, characterized by sudden family death with recessive inheritance. In the early 1960’s, the subtype without congenital deafness, characterized by prolongation of the QT interval, with syncope history and sudden death with autosomal dominant inheritance, was described independently by both an Italian pediatrician and an Irish pediatrician, resulting in this disease being called Romano-Ward syndrome. In 1979, the Prospective International Registry for Long QT Syndrome was established, which enabled the evaluation of many families affected by the disease worldwide, enabling them to obtain fundamental information on the clinical history of the disease, its therapy, and its genetic basis (Moss AJ, Circulation 1991).
What are the causes of Long QT Syndrome?
Today we know that LQTS is caused by mutations in genes that control potassium and sodium currents. Currently at least 15 involved genes have been identified, of which the three most important are: 1) KCNQ1, which results in LQT1, 2) KCNH2 (HERG), which results in LQT2, and 3) SCN5A, which results in LQT3. The anomalies in the potassium and / or sodium channels result in a ventricular repolarization defect, which is visualized on the ECG trace as prolongation of the QT interval and as morphological anomalies of ventricular repolarization, which favor the genesis of particular polymorphic ventricular tachycardias, called torsades-de-pointes, that can resolve spontaneously or degenerate into ventricular fibrillation.
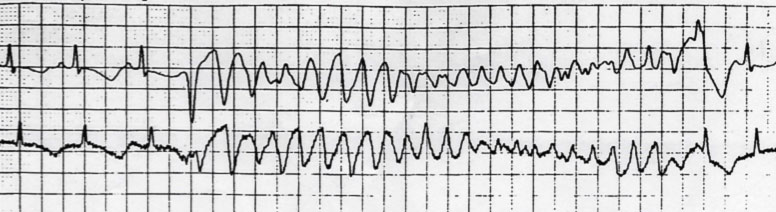
What is the incidence of Long QT Syndrome?
The prevalence of LQTS is estimated at 1:2,500 live births, with a reasonable variability in the different geographical areas. Most people with this condition develop symptoms before age 40. The age of onset of symptoms appears to differ based upon gender and the type of genetic variant. LQTS is a relatively common cause of sudden death, along with Brugada Syndrome and arrhythmogenic right ventricular dysplasia (ARVD), and in particular it could be responsible for some cases of Sudden Infant Death Syndrome (SIDS).
What are the symptoms of Long QT Syndrome?
Patients, mostly children and adolescents, present to the clinic following unexplained fainting (syncope), a family history of sudden death, or referral by a physician after a physical exam for participation in sports, during which the prolongation of the QT interval was observed.
The diagnosis can also occur in adulthood, particularly in female patients in post-partum or menopause, or following taking drugs that prolong the QT interval.
The clinical manifestations of LQTS are precisely linked to the appearance of episodes of polymorphic ventricular tachycardia called “Torsades de pointes”. The duration of the episodes determines the symptoms, ranging from syncope (i.e. transient loss of consciousness) to cardiac arrest to sudden death, when Torsades de Pointes degenerate into ventricular fibrillation.
Often the syncopal episodes of LQTS are mistaken for convulsive or epileptic seizures, thus delaying the diagnosis and the correct therapy of the disease. Collecting the patient’s family history, including episodes of fainting or convulsions, diagnosis of epilepsy or cases of sudden death at a young age, help to direct the diagnosis.
What are the electrocardiographic features of Long QT Syndrome?
The diagnosis of LQTS is made mainly by the electrocardiographic tracing, using either 12-derivation ECG or dynamic Holter ECG, with the detection of the prolongation of the QT interval corrected for the heart rate according to the Bazett formula.
In patients with a history of syncope, the diagnosis of long QT is made when the duration of the corrected QT interval is greater than 460 msec. In the absence of syncope or family history of LQTS or documented arrhythmias, the clinical diagnosis of LQTS may require the presence of a corrected QT interval greater than or equal to 480 msec.
It should be noted that the prolongation of the QT interval may not be present in the resting ECG trace, but may be documented during dynamic ECG Holter monitoring.
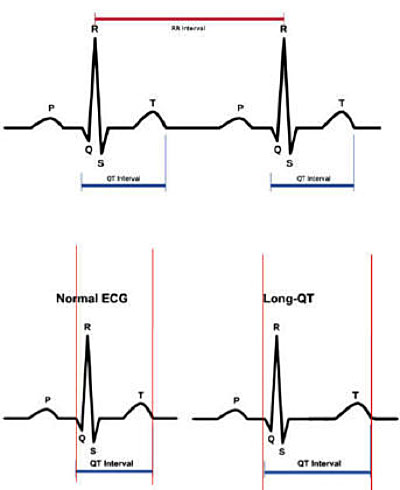
For the correction of the QT interval for heart rate, in addition to the Bazett formula, there is also the Fredericia formula, particularly used in pediatric patients, and in particular in the analysis of the QT interval during Holter recordings, since the Bazett formula can overestimate the duration of the QT interval in case of marked tachycardia.
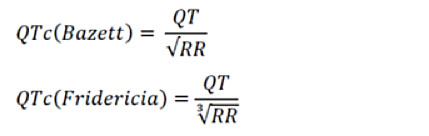
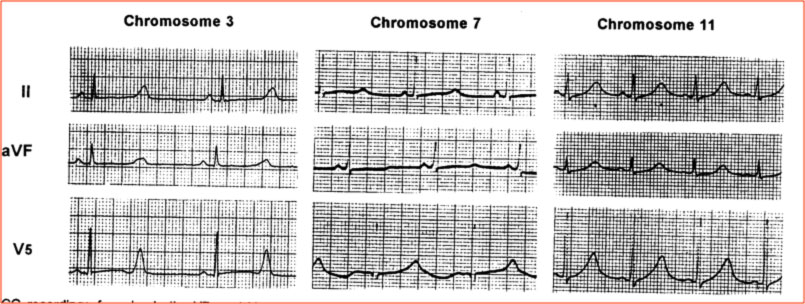
Nella LQTS, oltre che la durata dell’intervallo QT, è alterata anche la morfologia dell’onda T, con la comparsa di particolari anomalie, dette incisure o notches. La morfologia dell’ECG è stato descritto essere differente nelle diverse varianti genetiche (Moss, Circulation 1991).
What are the genetic characteristics of Long QT Syndrome?
Long QT Syndrome is caused by mutations in genes that control potassium and sodium currents. Currently at least 15 involved genes have been identified, of which the three most important are: 1) KCNQ1, which results in LQT1, 2) KCNH2 (HERG), which results in LQT2, and 3) SCN5A, which results in LQT3. In familial forms, pathogenic variants of these genes are identified in a percentage of patients ranging from 60% to 75% of cases. Other genetic variants are identified in very low percentages (about 1-2% of cases), while in about 20% of cases no known genetic mutations are identified.
| Genotype | Gene | Chromosome | Incidence | Current |
| LQT1 | KCNQ1 | 11 | 30-35% | Iks (reduced) |
| LQT2 | KCNH2 | 7 | 25-30% | Ikr (reduced) |
| LQT3 | SCN5A | 3 | 5-10% | Ina (increased) |
Genetic analysis is therefore fundamental in the diagnosis of LQTS, and has become an essential component for clinical management. The identification of the gene that causes the disease makes it possible to carry out a family screening, to quickly identify the family members carrying the mutation, thus allowing the prevention of sudden death in the whole family.
However, numerous recent studies indicate that LQTS cannot always be explained by a single genetic mutation, but seems to follow a more complex genetic model, where common genetic polymorphisms could have a specific effect on disease expression (modifier genes).
Finally, many studies are already in place to develop gene-specific treatments in LQTS, while a gene-specific prevention of the risk of events is already possible, since the triggers of the events are different in the various genetic forms.
In our center, the analysis of long QT genes is carried out by means of a “Next Generation Sequencing” (NGS) platform that enables the simultaneous study of more patients for mutations in different genes. All variants identified by NGS are always confirmed with another sequencing method, Sanger sequencing.
If you want to learn more about our Cardiogenetic Service click here.
What are the triggers of arrhythmias in Long QT Syndrome?
There is a close correlation between the factors that trigger arrhythmias and the genetic variant of which the patient is a carrier. Therefore, the prevention of the risk of arrhythmias is different depending on the genetic form.
In LQT1 patients, the largest genetic subgroup, most life-threatening cardiac events occur during exercise, particularly during swimming. Patients with LQT1 are also sensitive to potassium depletion.
LQT2 patients are particularly sensitive to emotions and sudden noises, such as the ringing of the telephone or that of the alarm clock, and moreover, the LQT2 females seem to be at higher risk in the postpartum period. Patients with LQT2 are also the most exposed to the risk of proarrhythmic effects of some drugs, which act as blockers of the Ikr current, and of potassium depletion.
LQT3 patients have more frequent events during resting or sleeping conditions. The pivotal therapy is β-blocker therapy, which has shown efficacy in reducing mortality in the LQTS patient population.
What are the therapies of Long QT Syndrome?
Patients with LQTS should always avoid drugs that prolong the QT interval (www.crediblemeds.org). In patients with LQTS, it is important to correct electrolyte disorders (hypokalemia, hypomagnesaemia, hypocalcemia) that may occur in the case of metabolic or gastrointestinal illness problems, and in the case of diuretic therapies.
First choice drug therapy is based on the use of full-dose beta-blockers. Some studies show that, in LQTS, not all β-blockers have equal efficacy and that the most effective beta-blockers are propranolol and nadolol.
The use of beta-blockers is recommended in all subjects with a clinical diagnosis of LQTS, even if asymptomatic, given that beta-blockers reduce mortality from 60% to less than 2%.
The implantable cardioverter-defibrillator (ICD) is recommended in patients with previous cardiac arrest. ICD implantation is also recommended when a patient receiving full-dose beta-blockers has a new syncope episode. The ICD is also recommended in asymptomatic patients with a very prolonged QT (QTc> 500 msec), carrying a pathogenic mutation in one of the three main genes.
In patients with an ICD who have an arrhythmic recurrence, or in patients with contraindications to beta-blockers or ICDs, the execution of left sympathetic cardiac denervation can be considered.
Sodium channel blockers (Mexiletine or Flecainide or Ranolazine) can be considered as additive therapy to shorten the QT interval in LQT3 patients with QTc greater than 500 msec.
Essential bibliography
- Moss AJ, Schwartz PJ, Crampton RS, Tzivoni D, Locati EH, MacCluer J, Hall WJ, Weitkamp L, Vincent GM, Garson A, Jr. and et al. The long QT syndrome. Prospective longitudinal study of 328 families. Circulation 1991; 84:1136-1144.
- Priori, SG, Wilde AA, Horie M et al: HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndrome. Heart rhythm 2013; 10: 287-294.
- Moss AJ, Zareba W, Benhorin J, Locati EH, Hall WJ, Robinson JL, Schwartz PJ, Towbin JA, Vincent GM, Lehmann M, Keating MT, MacCluer JW, Timothy KW: ECG T wave patterns in genetically distinct forms of the hereditary long QT syndrome. Circulation 1995; 92: 2929-2934.
- Schwartz PJ, Priori SG, Locati EH, Napolitano C, Cantu’ F, Towbin AJ, Keating MT, Hammoude H, Brown AM, Chen LK, Cotasky TJ: Long QT syndrome patients with mutations on the SCN5A and HERG genes have differential responses to Na+ channel blockade and to increase in heart rate. Implications for gene-specific therapy. Circulation 1995; 92: 3381-86.
- Mizusawa Y, Horie M, Wilde AAM, Genetic and Clinical Advances in Congenital Long QT Syndrome., Circ J 2014; 78: 2827–2833.