Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia (ARVC/D)
Indice dell'articolo
What is arrhythmogenic right ventricular dysplasia (ARVD), or arrhythmogenic right ventricular cardiomyopathy (ARVC)?
Arrhythmogenic right ventricular dysplasia (ARVD) is a cardiac muscle disease, clinically characterized by potentially lethal ventricular arrhythmias. The prevalence is estimated at 1:2,000, although in some countries (Italy and Greece) the disease is particularly common (as much as 1:700). The disease consists of a degeneration of the ventricular myocardium, mainly localized to the right ventricle, but which may also involve the left ventricle. The cardiac muscle tissue (called myocardium) is replaced by a fibro-adipose tissue of such magnitude as to cause aneurysms (dilations) of the ventricle. It is not easy to estimate the true prevalence and incidence of ARVD because patients are not always easily identifiable from a diagnostic point of view. Furthermore, sometimes the first manifestation of the pathology is sudden cardiac death, and this complicates epidemiological investigations. The prevalence of the disease is similar in both sexes, although the majority of symptomatic patients are male.
What is the clinical presentation of ARVD?
ARVD is one of the major causes of sudden death in young adults and athletes. Its clinical presentation usually consists of arrhythmic phenomena ranging from more or less frequent isolated ventricular extrasystoles to tachycardia or ventricular fibrillation (VT/VF).
What are the genetic characteristics of ARVD?
Based on current knowledge, ARVD has a genetic basis, and no acquired forms are known. Numerous genes have been described that, when mutated, cause ARVD. These include the genes PKP2, DSG2, DSC2, TGFB3, DSP, JUP, and TMEM43; more recently, the genes LDB3, LMNA, RYR2, TTN and CTNNA3 have also been added. The genes that cause the disease code for the proteins of mechanical cellular junctions (placoglobin, placofilina, desmoglein, desmocolina, desmoplakina).
A family history of ARVD is present in a percentage ranging from 30 to 50% of cases. The most common transmission pattern is autosomal dominant with variable penetrance and polymorphic phenotypic expression, although an autosomal recessive model has also been described. The most common hereditary modality of ARVD is of an autosomal dominant type: it is sufficient to possess a mutated single copy of a disease gene (mutation in heterozygosity) to be affected. In many families with a known ARVD genotype, incomplete penetrance (not all heterozygotes for causative mutations presenting clinical manifestations) and variable expressivity (the clinical picture may vary even between genetically related individuals and heterozygotes for the same causative mutation) were found. In addition to the typical form with autosomal dominant transmission and variable penetrance, recessive forms associated with palmo-plantar keratoderma and woolly hair have also been observed.
How is the ARVD diagnosis made?
The diagnosis of ARVD usually occurs during adolescence or adulthood. Because of the progressive evolution of the pathology, patients often present with a heterogeneous symptomatology. Palpitations, fatigue and syncope seem to be the most common symptoms, but sometimes there are non-specific symptoms, such as abdominal pain and mental confusion. In some cases, however, the first manifestation may be cardiac arrest in conjunction with intense physical effort, or at night. The main differential diagnoses are myocarditis, dilated cardiomyopathy, sarcoidosis and amyloidosis.
The diagnosis of ARVD begins with taking a good medical history. A personal history of palpitations (especially in young people), together with a family history of sudden death, should always lead to the suspicion of ARVD.
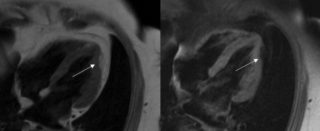
The tools of choice for diagnosis are imaging tests, including two-dimensional ultrasound, angiography and magnetic resonance imaging, which reveal structural and functional anomalies. The electroanatomical map enables the identification of the low voltage areas corresponding to myocardial atrophy, with fat-fibrous substitution.
What are the typical electrocardiographic abnormalities of ARVD?
50 to 90% of patients have electrocardiographic abnormalities, which include:
1) inversion of the T wave in precordial derivations, in subjects aged> 12 years and in the absence of RBBB
2) onda epsilon (low amplitude potentials located at the beginning of the ST segment – represent a delay in activation of right ventricular myocardium)
3) frequent ventricular extrasystoles (> 1000 BEV / 24 hours) with left bundle branch morphology,
4) unsupported ventricular tachycardias (NSVT) or sustained ventricular tachycardias (SVT) with left bundle branch morphology (although it is possible to find VT with different morphologies).
5) Late potentials on high resolution ECG
What are the risk factors for the ARVD prognosis?
The major risk factors for a poor prognosis are young age, family history of sudden juvenile death, a QRS greater than or equal to 140 ms, inversion of the T wave on the tracing, the extent of involvement of the right ventricle, the involvement of the left ventricle, the presence of ventricular tachycardias, the history of syncope or a previous cardiac arrest.
The endocavitary electrophysiological study (EES) is used for risk stratification in order to evaluate the susceptibility of the arrhythmogenic substrate. With the EES, it is possible to determine the type of inducible arrhythmia, as well as its morphology and hemodynamic tolerability.
What is the clinical evolution of ARVD?
The natural history of ARVD depends both on the electrical instability of the substrate and on the progressive ventricular dysfunction. With the evolution of the pathology, there is an alteration of the ventricular contractility that results in heart failure in either the right ventricule, or in both ventricules. The involvement of the left ventricle, whether it is macroscopic or histological, affects about 70% of patients. This involvement appears to be dependent on age and intense exercise (for example in competitive athletes), which seems to worsen and accelerate the clinical evolution of the disease. It is therefore recommended to follow the clinical evolution with serial echocardiographic investigations. In the natural progression, however, the following phases could be considered:
1) initial or occult phase, characterized by minimal structural alterations, with or without minor ventricular arrhythmias, during which sudden death could occasionally be the first manifestation of disease (particularly during intense physical activity);
2) arrhythmogenic phase, during which ventricular arrhythmias originating from the right ventricle are symptomatic and can lead to cardiac arrest (whether or not there are functional anomalies of the ventricle);
3) right ventricular dysfunction phase, with the progressive appearance of right ventricular failure due to the progressive replacement of the muscle with fibroadipose tissue, with relatively conserved left ventricular function;
4) phase of biventricular dysfunction, in which there is progressive biventricular involvement and dysfunction. At this stage, the ARVD can mimic a different dilated cardiomyopathy and lead to congestive heart failure with all its related complications, such as atrial fibrillation and thromboembolic events.
What is the therapy for ARVD?
The main treatment of arrhythmogenic dysplasia is aimed at preventing sudden cardiac death. Although there is no way to cure ARVD, it is possible to control arrhythmic manifestations and ventricular dysfunction. Available therapies include lifestyle modifications, antiarrhythmic drugs, ablation of the arrhythmic substrate with radiofrequency, and implantation of an automatic defibrillator (ICD).
Although there is no definitive evidence regarding lifestyle changes, patients should avoid intense physical activity, both to avoid arrhythmias and to avoid ventricular overload that can promote ventricular dysfunction.
In the case of arrhythmias, antiarrhythmic drugs can be used, which have also shown limited efficacy. Amiodarone, administered intravenously, is effective in interrupting ventricular tachycardias. Other pharmacological regimens include beta-blockers, either alone or in combination with class Ia and Ic drugs, amiodarone in combination with class II or Ic drugs.
Radiofrequency ablation is used in the case of incessant or refractory well-localized ventricular tachycardias, after the defibrillator is implanted to reduce its intervention. The effectiveness of ablation varies from case to case, and sometimes more than one procedure is required. Recurrences are often due to the evolution of the pathology that creates new re-entry circuits.
The implantation of a defibrillator, generally, is recommended for patients with previous resuscitated cardiac arrest, or who have induced sustained ventricular arrhythmias during EES, or with unsupported ventricular arrhythmias and history family of sudden death at a young age, or when there is involvement of the left ventricle.