Il tuo account è temporaneamente sospeso.
IL numero di assenze al corso determina la momentanea sospensione all’accesso ai contenuti riservati.
menu trigger
menu trigger

Richiedi AppuntamentoRichiedi Informazioni+ 39 02 52774260Piazza Edmondo Malan 2, San Donato M.se - Milano
admin5334, Autore presso AF-ABLATION

24 novembre 2022
Obiettivo del Convegno è l’approfondimento delle prospettive e problematiche della gestione multidisciplinare del paziente con fibrillazione atriale, in particolare l’individuazione e gestione di nuovi fattori di rischio, di nuove metodiche diagnostiche e nuove modalità terapeutiche mediche, elettriche e chirurgiche.
RESPONSABILI SCIENTIFICI
Prof. Giuseppe Ciconte
Prof. Emanuela T. Locati
Prof. Carlo Pappone
Per prendere visione del programma
![]()

Incontro del Rotary Club di Benevento con il Professore Carlo Pappone
– Ordinario di Cardiologia e Direttore della Cattedra di Cardiologia all’Università San Raffaele –
sul tema: “Prevenzione morte improvvisa”


Circulation. Arrhythmia and electrophysiology
2020 Sep;13(9):e008524. doi: 10.1161/CIRCEP.120.008524. Epub 2020 Jul 28.
PMID: 32755392 DOI: 10.1161/CIRCEP.120.008524
Peter M van Dam, Emanuela T Locati, Giuseppe Ciconte, Valeria Borrelli, Francesca Heilbron, Vincenzo Santinelli, Gabriele Vicedomini, Michelle M Monasky, Emanuele Micaglio, Luigi Giannelli, Valerio Mecarocci, Žarko Ćalović, Luigi Anastasia, Carlo Pappone
Abstract
Background: In Brugada syndrome (BrS), diagnosed in presence of a spontaneous or ajmaline-induced type-1 pattern, ventricular arrhythmias originate from the right ventricle outflow tract (RVOT). We developed a novel CineECG method, obtained by inverse electrocardiogram (ECG) from standard 12-lead ECG, to localize the electrical activity pathway in patients with BrS.
Methods: The CineECG enabled the temporospatial localization of the ECG waveforms, deriving the mean temporospatial isochrone from standard 12-lead ECG. The study sample included (1) 15 patients with spontaneous type-1 Brugada pattern, and (2) 18 patients with ajmaline-induced BrS (at baseline and after ajmaline), in whom epicardial potential duration maps were available; (3) 17 type-3 BrS pattern patients not showing type-1 BrS pattern after ajmaline (ajmaline-negative); (4) 47 normal subjects; (5) 18 patients with right bundle branch block (RBBB). According to CineECG algorithm, each ECG was classified as Normal, Brugada, RBBB, or Undetermined.
Results: In patients with spontaneous or ajmaline-induced BrS, CineECG localized the terminal mean temporospatial isochrone forces in the RVOT, congruent with the arrhythmogenic substrate location detected by epicardial potential duration maps. The RVOT location was never observed in normal, RBBB, or ajmaline-negative patients. In most patients with ajmaline-induced BrS (78%), the RVOT location was already evident at baseline. The CineECG classified all normal subjects and ajmaline-negative patients at baseline as Normal or Undetermined, all patients with RBBB as RBBB, whereas all patients with spontaneous and ajmaline-induced BrS as Brugada. Compared with standard 12-lead ECG, CineECG at baseline had a 100% positive predictive value and 81% negative predictive value in predicting ajmaline test results.
Conclusion: In patients with spontaneous and ajmaline-induced BrS, the CineECG localized the late QRS activity in the RVOT, a phenomenon never observed in normal, RBBB, or ajmaline-negative patients. The possibility to identify the RVOT as the location of the arrhythmogenic substrate by the noninvasive CineECG, based on the standard 12-lead ECG, opens new prospective for diagnosing patients with BrS.

La sindrome di Noonan è una malattia genetica pleomorfa a trasmissione autosomica dominante caratterizzata da una facies caratteristica, bassa statura, difetti cardiaci congeniti e ritardo dello sviluppo di grado variabile.
Nel 1968, la dott.ssa Jacqueline Noonan descrisse la sindrome omonima, raggruppando nove pazienti che mostravano una serie di caratteristiche cliniche simili associate a cardiopatie congenite più o meno complesse.
Queste caratteristiche, che possono essere più o meno pronunciate , includono una facies tipica ( ipertelorismo, ptosi , orecchie basse, forma triangolare del viso), bassa statura e anomalie del torace (pectus excavatum inferiore o pectus carinatum superiore).
Ulteriori caratteristiche extracardiache includono disabilità dello sviluppo neurologico, criptorchidismo, pubertà ritardata, linfedema, disturbi emorragici e una predisposizione leggermente aumentata a tumori ematologici e solidi (Roberts, Allanson, Tartaglia e Gelb, 2013).
È stato stabilito che la trasmissione sia autosomica dominante, sebbene sia stata recentemente identificata una forma autosomica recessiva (Johnston et al., 2018). Il fenotipo è variabile, ma si ritiene che la penetranza sia completa.
La sindrome di Noonan condivide molti aspetti con altre sindromi quali la sindrome di Costello, sindrome cardiofaciocutanea, la sindrome di Noonan con lentiggini multiple (precedentemente nota come sindrome LEOPARD).
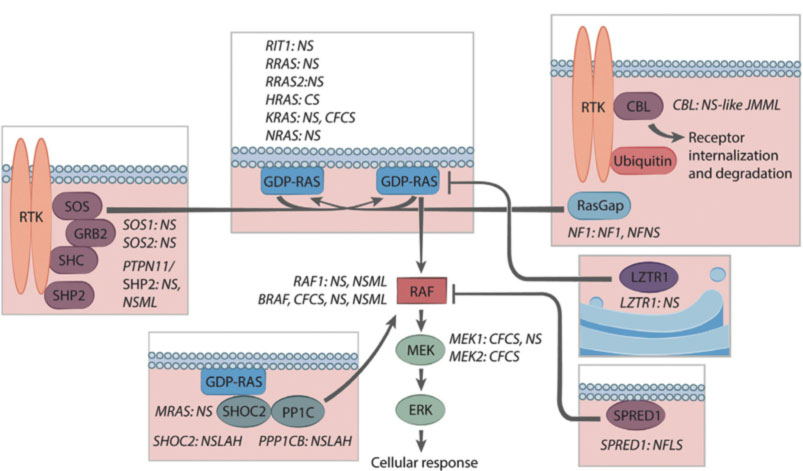
Tali patologie sono contraddistinte da una medesima fisiopatologia secondaria a mutazione dei geni nella via del signaling RAS/MAPK. Nell’insieme esse costituiscono le così dette Rasopatie.
Dal 2001, quando è stato scoperto il PTPN11, il primo gene identificato per la Sindrome di Noonan (Tartaglia et al., 2001), il grado di eterogeneità molecolare alla base delle RASopatie è diventato evidente.

Una delle principali caratteristiche della NS, identificata fin dalla prima descrizione, è il coinvolgimento cardiaco.
I pazienti affetti presentano un ampio spettro di malattie cardiache, che rientrano in due categorie principali: cardiopatie congenite (CHD), presente in circa l’80% dei pazienti, e cardiomiopatia ipertrofica (HCM), riscontrata in circa il 20% dei pazienti (Marino et al., 1999).
La cardiopatia congenita si manifesta molto spesso come stenosi della valvola polmonare (PVS), con una prevalenza stimata di ~ 40%, ma sono stati descritti altri tipi, in particolare difetti del setto interatriale (8%) o interventricolare (ASD / VSD), nonché difetti del canale atrioventricolare (AVCD) (15%) (Calcagni et al., 2017).
Meno frequentemente, i pazienti presentano forme di cardiopatia che interessano le strutture cardiache sinistre tra cui la stenosi della valvola mitrale (6%), stenosi della valvola aortica (AS) e coartazione aortica (9%) (Lam, Corno, Oorthuys e Marcelletti, 1982).
Dopo la delucidazione delle varianti genetiche responsabili della SN, è diventato possibile cercare diverse associazioni genotipo-fenotipo. Ne sono emerse in discreto numero anche se alcune associazioni sono lontane dall’esser chiaramente identificate a causa dell’esiguità del numero dei pazienti.
Le varianti patogene del PTPN11, ad esempio, costituiscono quasi l’80% delle cause identificate di NS tra i pazienti che presentano stenosi della valvola polmonare o difetti interatriali (Prendiville et al., 2014); al contrario, le mutazioni a carico di PTPN11 sono negativamente associate con la cardiopatia ipertrofica HCM (6% vs 26% dei pazienti afetti non PTPN11) (Tartaglia et al., 2002).
Le varianti patogene del gene RAF1 sono fortemente associate alla miocardiopatia ipertrofica ma negativamente associate a PVS (Pandit et al., 2007).
La variazione patogena in RIT1 predispone i pazienti ad anomalie valvolari che alla cardiopatia ipertrofica
La patogenesi delle CHD nella sindrome di Noonan , in particolare per quanto riguarda l’eterogeneità dei difetti cardiaci è solo parzialmente compresa. Per il prodotto del gene PTPN11, la proteina tirosina fosfatasi SHP2, è stato stabilito un chiaro ruolo nella morfogenesi valvolare.
In particolare, l’attivazione della via MAPK attraverso i recettori del fattore di crescita epidermico costituisce il meccanismo principale che guida la transizione epitelio-mesenchimale (EMT) nella formazione della valvola semilunare (Krenz, Yutzey e Robbins, 2005).
Clinical Case
Nel novembre del 2020 giungeva presso il pronto soccorso del nostro istituto un ragazzo di 20 anni per un episodio sincopale intervenuto a riposo.
La TAC encefalo e l’esame neurologico non mostravano alcuna acuzie e pertanto veniva indirizzato alla nostra attenzione.
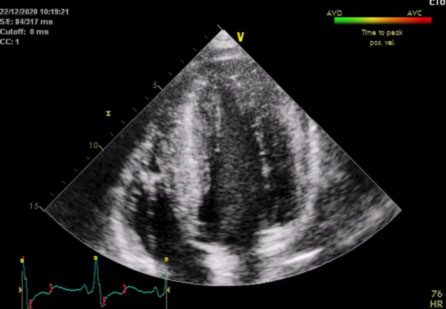
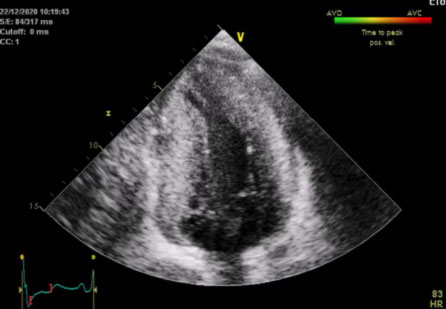
L’ECG ottenuto all’ingresso mostrava chiari segni di ipertrofia ventricolare sinistra. Per tale motivo veniva richiesto anche un approfondimento mediante ecocardiografia.
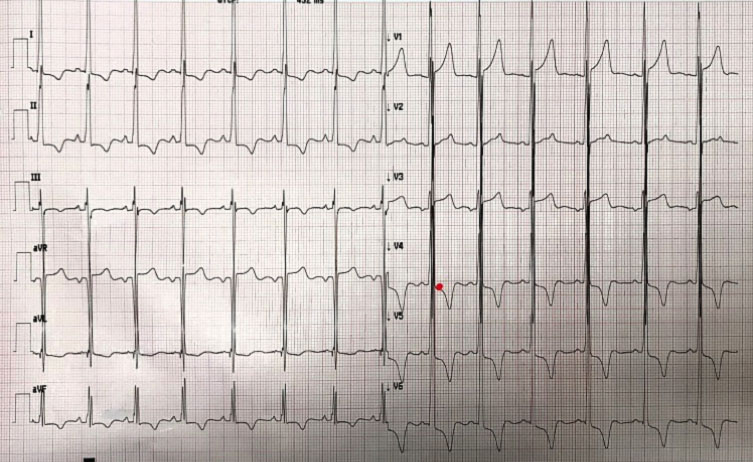
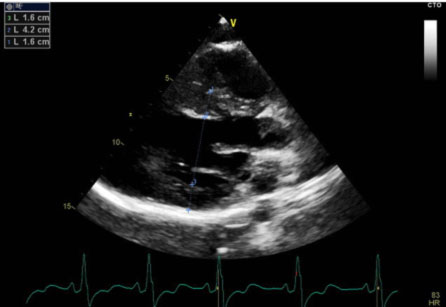
L’ecocardiografia mostrava i seguenti parametri: Severa ipertrofia concentrica del ventricolo sinistro con trabecolatura dei segmenti medio apicali, con ipertrofia a distribuzione asimmetrica prevalentemente localizzata a livello del setto interventricolare inferiore (21 mm) e della parete inferiore. La parete posteriore mostra spessore di ca. 16 mm. Funzione sistolica del ventricolo sinistro ai limiti inferiori di norma (FE 50%). Non segni di ostruzione all’efflusso sinistro. Pattern diastolico da alterato rilasciamento con normali pressioni di riempimento (E/e’ < 10). Atrio sinistro non dilatato. Radice aortica ed aorta ascendente di normali dimensioni. Ventricolo destro non dilatato, con lieve deficit della funzione sistolica (TAPSE 16 mm). Vena cava inferiore non dilatata e con normali escursioni respiratorie.
Il quadro ecocardiografico era pertanto compatibile con la diagnosi di miocardiopatia ipertrofica non ostruttiva.
 |
 |
 |
 |
Tale riscontro rendeva suggestiva l’ipotesi di una sincope a genesi aritmica. Per tale motivo si disponeva ricovero al fine di eseguire ulteriori accertamenti diagnostici.
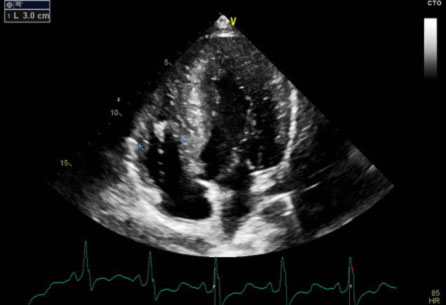
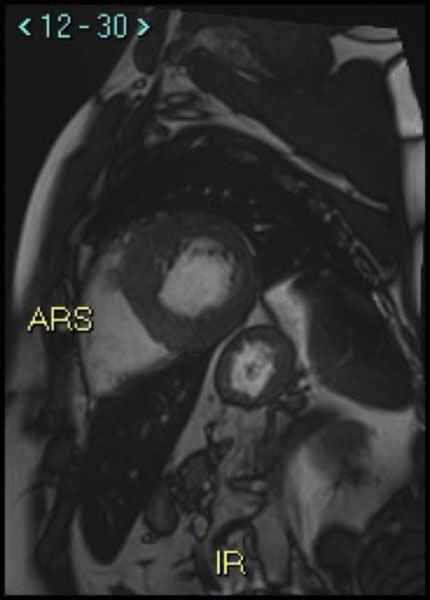
La risonanza magnetica cardiaca (CMRI) con mezzo di contrasto mostrava quadro di miocardiopatia ipertrofica con estesa fibrosi miocardica localizzata in prossimità delle parete inferosettali, anteriore ed anterolaterale (LGE> 30% della massa miocardica totale).
 |
 |
La presenza di una così vasta area di fibrosi miocardica pone il paziente ad un aumentato rischio di morte cardiaca improvvisa secondaria ad aritmie ventricolari sostenute (fibrillazione ventricolare o tachicardia ventricolare).
In considerazione dell’episodio sincopale si decideva pertanto di impiantare un defibrillatore in prevenzione primaria.
Ad un’esame fisico attento inoltre il paziente mostrava alcune caratteristiche peculiari : Bassa statura , fronte ampia, solchi naso genieni molto evidenti.
La facies complessiva suggeriva un’aspetto sindromico compatibile con la sindrome di Noonan.
Richiesta quindi la consulenza del collega genetista si procedeva anche ad espletare i relativi test genetici.
I test genetici mostravano una mutazione in eterozigosi a carico del gene SOS1 (Ras/Rac guanine nucleotide exchange factor 1).

Frontiers in pharmacology
2021 Apr 30;12:651720. doi: 10.3389/fphar.2021.651720. eCollection 2021.
PMID: 33995067 PMCID: PMC8120428 DOI: 10.3389/fphar.2021.651720
Emanuele Micaglio, Emanuela T Locati, Michelle M Monasky, Federico Romani, Francesca Heilbron, Carlo Pappone
Abstract
Adverse drug reactions (ADRs) are an important and frequent cause of morbidity and mortality. ADR can be related to a variety of drugs, including anticonvulsants, anaesthetics, antibiotics, antiretroviral, anticancer, and antiarrhythmics, and can involve every organ or apparatus. The causes of ADRs are still poorly understood due to their clinical heterogeneity and complexity. In this scenario, genetic predisposition toward ADRs is an emerging issue, not only in anticancer chemotherapy, but also in many other fields of medicine, including hemolytic anemia due to glucose-6-phosphate dehydrogenase (G6PD) deficiency, aplastic anemia, porphyria, malignant hyperthermia, epidermal tissue necrosis (Lyell’s Syndrome and Stevens-Johnson Syndrome), epilepsy, thyroid diseases, diabetes, Long QT and Brugada Syndromes. The role of genetic mutations in the ADRs pathogenesis has been shown either for dose-dependent or for dose-independent reactions. In this review, we present an update of the genetic background of ADRs, with phenotypic manifestations involving blood, muscles, heart, thyroid, liver, and skin disorders. This review aims to illustrate the growing usefulness of genetics both to prevent ADRs and to optimize the safe therapeutic use of many common drugs. In this prospective, ADRs could become an untoward “stress test,” leading to new diagnosis of genetic-determined diseases. Thus, the wider use of pharmacogenetic testing in the work-up of ADRs will lead to new clinical diagnosis of previously unsuspected diseases and to improved safety and efficacy of therapies. Improving the genotype-phenotype correlation through new lab techniques and implementation of artificial intelligence in the future may lead to personalized medicine, able to predict ADR and consequently to choose the appropriate compound and dosage for each patient.
Copyright © 2021 Micaglio, Locati, Monasky, Romani, Heilbron and Pappone.

Frontiers in cardiovascular medicine
2021 Apr 21;8:652027. doi: 10.3389/fcvm.2021.652027. eCollection 2021.
PMID: 33969014 PMCID: PMC8096997 DOI: 10.3389/fcvm.2021.652027
Michelle M Monasky, Emanuele Micaglio, Emanuela T Locati, Carlo Pappone
Abstract
The evolution of the current dogma surrounding Brugada syndrome (BrS) has led to a significant debate about the real usefulness of genetic testing in this syndrome. Since BrS is defined by a particular electrocardiogram (ECG) pattern, after ruling out certain possible causes, this disease has come to be defined more for what it is not than for what it is.
Extensive research is required to understand the effects of specific individual variants, including modifiers, rather than necessarily grouping together, for example, “all SCN5A variants” when trying to determine genotype-phenotype relationships, because not all variants within a particular gene act similarly.
Genetic testing, including whole exome or whole genome testing, and family segregation analysis should always be performed when possible, as this is necessary to advance our understanding of the genetics of this condition. All considered, BrS should no longer be considered a pure autosomal dominant disorder, but an oligogenic condition.
Less common patterns of inheritance, such as recessive, X-linked, or mitochondrial may exist. Genetic testing, in our opinion, should not be used for diagnostic purposes. However, variants in SCN5A can have a prognostic value. Patients should be diagnosed and treated per the current guidelines, after an arrhythmologic examination, based on the presence of the specific BrS ECG pattern.
The genotype characterization should come in a second stage, particularly in order to guide the familial diagnostic work-up. In families in which an SCN5A pathogenic variant is found, genetic testing could possibly contribute to the prognostic risk stratification.
Copyright © 2021 Monasky, Micaglio, Locati and Pappone.

European heart journal
2021 Mar 14;42(11):1082-1090. doi: 10.1093/eurheartj/ehaa942.
PMID: 33221895 PMCID: PMC7955973 DOI: 10.1093/eurheartj/ehaa942
Giuseppe Ciconte, Michelle M Monasky, Vincenzo Santinelli, Emanuele Micaglio, Gabriele Vicedomini, Luigi Anastasia, Gabriele Negro, Valeria Borrelli, Luigi Giannelli, Francesca Santini, Carlo de Innocentiis, Roberto Rondine, Emanuela T Locati, Andrea Bernardini, Beniamino C Mazza, Valerio Mecarocci, Žarko Ćalović, Andrea Ghiroldi, Sara D’Imperio, Sara Benedetti, Chiara Di Resta, Ilaria Rivolta, Giorgio Casari, Enrico Petretto, Carlo Pappone
Abstract
Aims: Brugada syndrome (BrS) is associated with an increased risk of sudden cardiac death due to ventricular tachycardia/fibrillation (VT/VF) in young, otherwise healthy individuals. Despite SCN5A being the most commonly known mutated gene to date, the genotype-phenotype relationship is poorly understood and remains uncertain. This study aimed to elucidate the genotype-phenotype correlation in BrS.
Methods and results: Brugada syndrome probands deemed at high risk of future arrhythmic events underwent genetic testing and phenotype characterization by the means of epicardial arrhythmogenic substrate (AS) mapping, and were divided into two groups according to the presence or absence of SCN5A mutation. Two-hundred probands (160 males, 80%; mean age 42.6 ± 12.2 years) were included in this study. Patients harbouring SCN5A mutations exhibited a spontaneous type 1 pattern and experienced aborted cardiac arrest or spontaneous VT/VF more frequently than the other subjects. SCN5A-positive patients exhibited a larger epicardial AS area, more prolonged electrograms and more frequently observed non-invasive late potentials. The presence of an SCN5A mutation explained >26% of the variation in the epicardial AS area and was the strongest predictor of a large epicardial area.
Conclusion: In BrS, the genetic background is the main determinant for the extent of the electrophysiological abnormalities. SCN5A mutation carriers exhibit more pronounced epicardial electrical abnormalities and a more aggressive clinical presentation. These results contribute to the understanding of the genetic determinants of the BrS phenotypic expression and provide possible explanations for the varying degrees of disease expression.



Questo breve video racconta di una storia accaduta ad una famiglia nelle Marche.
La fortuna di un padre sopravvissuto ad un arresto cardiaco grazie all’intuito di un figlio, e la fortuna dei figli che grazie ad una diagnosi precoce hanno potuto affrontare con maggior cognizione e serenità la sindrome di Brugada.
