Introduzione
La displasia o cardiomiopatia aritmogena del ventricolo destro (ARVD/C), è una patologia del muscolo cardiaco caratterizzata da anomalie funzionali e strutturali dovute alla sostituzione del miocardio con tessuto adiposo o fibroadiposo. La presentazione clinica dell’ARVD solitamente consiste in fenomeni aritmici che variano dalla semplice extrasistolia ventricolare alla tachicardia o fibrillazione ventricolare (TV/FV) . Lo spettro di presentazione è comunque molto variabile e può includere anche alterazioni globali o segmentali del Ventricolo destro (Vdx,), alterazioni ECGrafiche della depolarizzazione/ripolarizzazione (visibili soprattuttonelle derivazioni precordiali destre), evoluzione in scompenso cardiaco destro o biventricolare che può mimare una cardiomiopatia dilatativa di altra origine . Da quando è stata descritta per la prima volta (circa 30 anni fa) come “auricolarizzzazione del Vdx, sono stati compiuti molti progressi nella comprensione delle manifestazioni cliniche, della patogenesi e della anatomia patologica.
Istopatologia
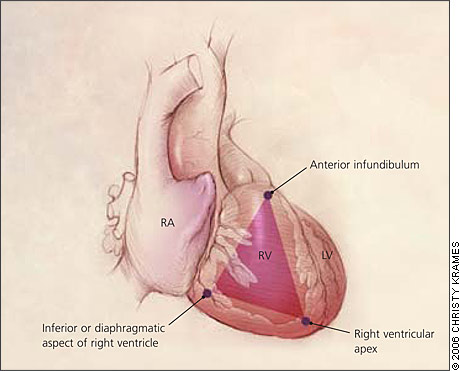
La caratteristica principale dell’ARVD è la sostituzione del miocardio a livello della parete libera del Vdx con tessuto adiposo o fibroadiposo. Solo i foglietti subendocardici appaiono più o meno preservati; a livello degli strati epicardici e mediomurali, all’interno dell’adipe, si osservano filoni di cardiomiociti delimitati o inglobati da aree variabili di fibrosi . In 2/3 dei casi è possibile riscontrare aree di miocardite acuta all’interno delle quali si riscontrano miociti morti ed un infiltrato infiammatorio (soprattutto linfociti). Questa patologia spesso viene trascurata in sede autoptica probabilmente perché il confine tra infarcimento adiposo patologico e fisiologico è molto labile. Si riconoscono 2 forme istopatologiche: la forma adiposa pura è caratterizzata da parziale o totale sostituzione del tessuto muscolare cardiaco con tessuto adiposo, con prevalente interessamento del’apice cardiaco e dell’infundibolo in assenza di fibrosi o infiltrati infiammatori. In questa variante il rischio di morte cardiaca improvvisa in assenza di altre coopatologie cardiache è controversa. La forma fibroadiposa ( la prima ad essere stata descritta) è caratterizzata da 1) tessuto fibroso che avvolge i cardiomiociti,2) dall’assottigliamento della parete del Vdx che spesso residua in una evoluzione aneurismatica, 3) dalla presenza di infiltrato infiammatorio. Gli aneurismi solitamente interessano le porzioni d’afflusso, d’efflusso ed apicali del Vdx (triangolo della displasia). In questa variante (sebbene in proporzioni minori) può venire interessato anche il ventricolo sinistro ed il setto.
Genetica
Una storia familiare di ARVD è presente in una percentuale che va dal 30 al 50% dei casi. Il più comune pattern di trasmissione è autosomico dominante con penetranza variabile ed espressione fenotipica polimorfica, sebbene sia stato anche descritto un modello autosomico recessivo. Le analisi di Linkage hanno localizzato le anomalie genetiche a livello dei cromosomi 1,2,3,14 per le forme dominanti e del cromosoma 17 per la forma recessiva . Quest’ultima forma è caratteristicamente associata alla malattia di Naxos. In questa condizione la patologia sembra avere un decorso più severo e la penetranza nei membri familiari si attesta al 90%.
E’ molto difficile stimare la reale prevalenza ed incidenza dell’ ARVD e questo perché i pazienti non sempre sono facilmente inquadrabili dal punto di vista diagnostico. Inoltre avvolte la prima manifestazione di questa è la morte cardiaca improvvisa e questo complica le indagini epidemiologiche.
Diagnosi
La diagnosi di ARVD solitamente viene fattta in età giovanile o adulta; la maggioranza dei pazienti affetti è di sesso maschile(M/F 3/1). A causa dell’evoluzione progressiva della patologia, i pazienti spesso presentano un corredo sintomatologico eterogeneo. Palpitazioni, fatica e sincope, sembrano essere i sintomi più comuni, ma talora sono presenti disturbi aspecifici come dolore addominale e confusione mentale. In alcuni casi laprima manifestazione può essere un arresto cardiaco in concomitanza con sforzi fisici intensi.
Segni e sintomi di ARVD
| Sintomi |
| Dolori addominali |
| Ridotta tolleranza allo sforzo |
| Confusione |
| Dispnea (specialmente sotto sforzo) |
| Fatica |
| Vertigini |
| Palpitazioni |
| Sincope |
| Segni |
| Arresto cardiaco |
| Edema periferico |
| Morte improvvisa |
| Tachicardia |
Lo diagnosi dell’ARVD comincia con una buona annotazione anamnestica. Una storia personale di palpitazioni (specie in persone di giovane età), con familiarità per morte improvvisa dovrebbe sempre indurre al sospetto della patologia in questione.
L’esame obiettivo risulta normale in almeno il 50% dei casi. Un importante ritrovamento diagnostico è uno sdoppiamento del secondo tono. Talora è possibile riscontrare anche un terzo ed un quarto tono. Raramente sono apprezzabili dei soffi. Se il ventricolo destro è molto dilatato invece, è possibile notare una asimmetria della parete toracica.
Dal 50 al 90% dei pazienti hanno delle peculiarità all’ECG di superfice. Queste peculiarità includono: 1) inversione dell’onda T nelle derivazioni precordiali, 2) onda epsilon,3) extrasistoli con morfologia a blocco di branca sinistro, 4) tachicardia ventricolare (TV) con morfologia a blocco di branca sinistro (anche se è possibile riscontrare TV con morfologie differenti).

Nel 1994 è stato istituito uno score system per fare diagnosi di ARVD che comprende dei criteri minori e dei criteri maggiori. Sebbene questi criteri siano specifici non sono dotati di buona sensibilità e non sono mai stati validati. Il criterio più affidabile per fare diagnosi resta quello istologico. Sfortunatamente però anche la biopsia manca di sufficiente sensibilità e questo a causa della natura segmentale della patologia e della tecnica stessa che va a prelevare il materiale bioptico dalla parete settale del ventricolo (difficilmente interessata dalla patologia). Nel sospetto di displasia aritmogena del ventricolo destro, il primo inquadramento diagnostico dovrebbe includere esami non invasivi come il monitoraggio ECG sec Holter, il test da sforzo,l’ecocardiogramma e la risonanza magnetica nucleare. Spesso tuttavia per una buona stratificazione del rischio si rendono necessari esami invasivi come la biopsia endomiocardica, l’angiografia ventricolare e lo studio elettrofisiologico.
Criteri Diagnostici per ARVD/C
| � Per la diagnosi di ARVD è necessaria la contemporanea presenza di :• 2 criteri maggiori oppure • 1 criterio maggiore e 2 minori oppure • 4 criteri minori
|
1 Riscontrate all’ Ecocardiogramma, RM, angiografia, scintigrafia.
E’ ancora materia di discussione quale sia la metodica di imaging più appropriata per la diagnosi. L’ecocardiografia con contrasto e l’angiografia possono identificare formazioni aneurismatiche, ed aree di discinesia nel triangolo della displasia ma sono tecniche invasive. La Risonanza magnetica nucleare al contrario è un esame non invasivo che consente una buona valutazione delle alterazioni strutturali e funzionali nonché una caratterizzazione indiretta del tessuto. Lo studio elettrofisiologico è utilizzato per una corretta stratificazione del rischio al fine di valutare la suscettibilità del substrato aritmogeno. Con lo S.E.F. è possibile determinare il tipo di aritmia inducibile, la sua morfologia e se essa sia o no emodinamìcamente tollerabile. Rimane inoltre un utile sussidio al fine della distinzione tra le aritmie idiopatiche del ventricolo destro che tendono ad avere un decorso benigno e la ARVD.
Storia naturale
La storia naturale dell’ARVD dipende sia dall’istabilità elettrica del substrato sia dalla progressiva disfunzione ventricolare. Come abbiamo già evidenziato le aritmie variano da semplici battiti ectopici ventricolari (con morfologia a blocco di branca sinistro) alla tachicardia ventricolare sostenuta o non sostenuta che possono evolvere in fibrillazione ventricolare e quindi in arresto cardiaco. E’ stato documentato uno squilibrio nell’innervazione adrenergica che potrebbe contribuire all’aritmogenesi a causa della dispersione del periodo refrattario e della generazione delle post depolarizzazioni tardive in particolare in concomitanza con l’esercizio fisico o l’aumento del tono catecolaminergico. Con l’evoluzione della patologia si assiste ad una alterazione della contrattilità ventricolare che residua in una insufficienza cardiaca destra o biventricolare. L’interessamento del ventricolo sinistro infatti, sia esso macroscopico o istologico, interessa circa il 76% dei pazienti affetti. Questo coinvolgimento sembra essere età dipendente, e più evidente in coloro che hanno una lunga storia di malattia con carattere di progressione come potrebbe essere valutato da indagini ecocardiografiche seriate. Nella storia naturale comunque potrebbero essere considerati i seguenti passaggi: 1) fase occulta, caratterizzata da alterazioni strutturali minime, con o senza aritmie ventricolari minori, durante la quale la morte improvvisa potrebbe essere occasionalmente la prima manifestazione di malattia (in particolare durante attività fisica intensa);2) fase di evidente disordine elettrico, durante la quale le aritmie ventricolari ad origine dal ventricolo destro sono sintomatiche e possono portare ad arresto cardiaco (in presenza o meno di anomalie funzionali del ventricolo); 3) Insufficienza del ventricolo destro dovuta alla progressiva sostituzione del muscolo con tessuto fibroadiposo con una funzione ventricolare sinistra relativamente conservata ; 4) lo stadio finale in cui si assiste ad un coinvolgimento biventricolare. In questa fase l’ARVD può mimare una diversa cardiomiopatia dilatativa e portare a scompenso cardiaco congestizio con tutte le sue relative complicanze quali fibrillazione atriale ed eventi tromboembolici.
Terapia
Il principale trattamento della displasia aritmogena è rivolto alla prevenzione della morte cardiaca improvvisa. Sebbene non ci sia modo di curare l’ARVD, le sue manifestazioni aritmiche o relative al dato funzionale, possono essere tenute sotto controllo. Le terapie disponibili includono modificazioni dello stile di vita, farmaci antiaritmici, ablazione con radiofrequenze, impianto di un defibrillatore automatico (AICD). Il primo passo nella gestione della patologia è informare il paziente circa la stessa. Sebbene non esistano evidenze definitive riguardo le modificazioni dello stile di vita, i pazienti dovrebbero evitare attività che possano scatenare episodi aritmici quali un’ eccessiva attività fisica. Una volta ottenuta una buona gestione delle aritmie, l’attività fisica potrebbe essere incrementtata sotto stretto controllo medico.
I farmaci antiaritmici sono comunemente utilizzati nella terapia della displasia aritmogena. Non esiste un singolo farmaco che abbia mostrato una completa efficacia, sebbene l’amiodarone somministrato per via endovenosa risulti efficacissimo nell’interrompere le tachicardie ventricolari. Altri regimi farmacologici comprendono antiaritmici di classe II (beta bloccanti come il propanololo), da soli o in associazione con farmaci di classe Ia (procainamide), farmaci di classe Ic, amiodarone in associazione a farmaci di classe II o Ic. L’ablazione mediante radiofrequenze è utilizzata in caso di 1)tachicardie ventricolari incessanti o refrattarie al trattamento medico, 1)tachicardie ventricolari ben localizzabili,3) successivamente all’impianto del defibrillatore per ridurre gli interventi dello stesso. L’obiettivo dell’ablazione è quello di distruggere quei pathway critici che portano al perpetuarsi della tachicardia. Il tasso di successo dell’ablazione varia da caso a caso ed avvolte è neccesaria più di una procedura; queste recidive spesso sono causa dell’evoluzione stessa della patologia che va a creare nuovi circuiti di rientro.
Impiantare un defibrillatore è una scelta che dipende soprattutto dall’ expertise locale e questo perché attualmente non esiste una adeguata stratificazione del rischio per questo tipo di patologia. In generale l’impianto del defibrillatore è raccomandato in coloro che 1) hanno aritmie refrattarie al trattamento medico,2) nei soggetti di giovane età, 3)in caso di pregresso arresto cardiaco resuscitato, 4)qualora ci sia coinvolgimento del ventricolo sinistro. Una delle poche controindicazioni all’impianto invece è costituita dalla tachicardia ventricolare incessante.
- Category
- Aritmie Adulti /Pediatrici