Sindrome del QT lungo (LQTS)
Indice dell'articolo
Che cos’è la sindrome del QT lungo?
La sindrome del QT lungo (LQTS) è una patologia cardiaca su base genetica, caratterizzata da un elevato rischio di aritmie cardiache ventricolari, che possono provocare sincope (perdita di coscienza), arresto cardiaco e morte improvvisa. La malattia è caratterizzata dal prolungamento dell’intervallo QT, misurato sull’elettrocardiogramma di superficie, e da anomalie morfologiche dell’onda T (descitte come onde T bifide op indentate, a “gobba di cammello”). La LQTS si manifesta prevalentemente in età pediatrica, ma in alcuni casi può esordire anche dopo la pubertà e in età adulta. La gravità della malattia è molto variabile e, almeno in parte, dipendente dal tipo di gene o di mutazione implicati.
La LQTS è stata riconosciuta per la prima volta da Jervell e Lange-Nielsen, nel 1957 in Norvegia, che descrissero la variante della sindrome associata a sordità neurologica, caratterizzata da morte improvvisa a carattere familiare con ereditarietà recessiva. Agli inizi degli anni sessanta la variante senza sordità congenita, caratterizzata da prolungamento dell’intervallo QT, con storia sincopi e morte improvvisa con ereditarietà autosomica dominante fu descritta indipendentemente da un pediatra italiano e da un pediatra irlandese, quindi tale malattia fu denominata sindrome di Romano-Ward.
Nel 1979 è stato istituito il Registro Prospettico Internazionale per la sindrome del QT lungo, che ha permesso di valutare numerose famiglie colpite dalla patologia a livello mondiale, permettendo di ottenere informazioni fondamentali sulla storia clinica della malattia, sulla sua terapia e sulle sue basi genetiche (Moss AJ, Circulation 1991).
Quali sono le cause della Sindrome del QT lungo?
Oggi sappiamo che la LQTS è causata da mutazioni su geni che controllano le correnti al potassio ed al sodio. Attualmente sono stati identificati almeno 15 geni coinvolti, di cui i tre più importanti sono: KCNQ1 che determina la variante LQT1, KCNH2 (HERG) che determina la variante LQT2, e SCN5A che determina la variante più rara LQT3. Le anomalie nei canali del potassio e/o del sodio determinano un difetto di ripolarizzazione ventricolare, che si visualizza sul tracciato ECG come prolungamento dell’intervallo QT e come anomalie morfologiche della ripolarizzazione ventricolare, che favoriscono la genesi di particolari tachicardie ventricolari polimorfe, dette torsione-di-punta (torsades-de-pointes), che possono risolversi spontaneamente oppure degenerare in fibrillazione ventricolare.
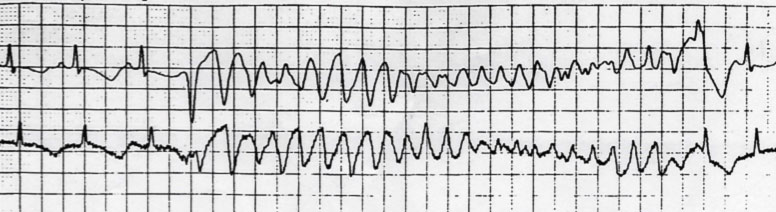
Esempio di Torsione di punta in paziente con LQTS
Qual è l’incidenza della sindrome del QT lungo?
La prevalenza della LQTS è stimata in 1 ogni 2.500 nati vivi, con una discreta variabilità nelle differenti aree geografiche. Nella LQTS è stata descritta una relativa prevalenza del sesso femminile, ancora non del tutto spiegata. La maggior parte dei soggetti con questa condizione sviluppa sintomi prima dei 40 anni, e l’età di comparsa dei sintomi sembra mostrare differenze in base al sesso e in base alle diverse varianti genetiche. La LQTS è una causa relativamente comune di morte improvvisa nel giovane (prima dei 40 anni di età), insieme alla Sindrome di Brugada (SdB)e alla Cardiomiopatia/displasia ventricolare destra aritmogena (CAVD), e in particolare potrebbe essere responsabile di una parte delle cosiddette “morti in culla” o SIDS (acronimo dall’inglese “Sudden Infant Death Syndrome”).
Quali sono i sintomi della sindrome del QT lungo?
I pazienti, per lo più bambini e adolescenti, giungono all’osservazione a seguito di svenimenti (sincopi) non attribuibili ad altre cause, o in seguito a storia familiare di morte improvvisa, o perché segnalati da medici sportivi che al momento della visita per l’idoneità notano il prolungamento dell’intervallo QT a livello elettrocardiografico.
La diagnosi può avvenire anche in età adulta, particolarmente in pazienti di sesso femminile particolarmente nella fase del post-partum, o nel climaterio, o a seguito dell’assunzione di farmaci che prolungano l’intervallo QT.
Le manifestazioni cliniche della LQTS sono appunto legate alla comparsa di episodi di tachicardia ventricolare polimorfa detta a “Torsione di Punta”. La durata degli episodi determina i sintomi che variano dalle sincopi (cioè perdite di coscienza transitorie) all’arresto cardiaco alla morte improvvisa quando le Torsioni di Punta degenerano in fibrillazione ventricolare.
Spesso gli episodi sincopali della LQTS vengono scambiati per crisi convulsive o epilettiche, ritardando così la diagnosi e la corretta terapia della malattia. Raccogliendo la storia familiare del paziente vengono spesso identificati episodi di svenimenti o convulsioni, diagnosi di epilessia o casi di morte improvvisa in giovane età, che indirizzano verso una malattia familiare e indirizzano quindi la diagnosi.
Quali sono le caratteristiche elettrocardiografiche della sindrome del QT lungo?
La diagnosi della LQTS avviene principalmente attraverso il tracciato elettrocardiografico, sia ECG 12 derivazioni che ECG dinamico Holter, con il rilevamento del prolungamento dell’intervallo QT, corretto per la frequenza cardiaca secondo la formula di Bazett.
In pazienti con storia di sincopi, la diagnosi di QT lungo viene effettuata quando la durata dell’intervallo QT corretto è maggiore di 460 msec. In assenza di sincope o di storia familiare di LQTS o di aritmie documentate, la clinica diagnosi di LQTS potrebbe richiedere la presenza di un intervallo QT corretto maggiore o uguale a 480 msec.
E’ da notare che il prolungamento dell’intervallo QT potrebbe non essere presente nel tracciato ECG a riposo, ma sessere documentato durante ECG dinamico secondo Holter. Inoltre all’interbo delle famiglie con LQTS sono stati individuati soggetti portatori della mutazione con intervallo QT nella norma. Pertanto la relazione tra diagnosi clinica di LQTS e durata dell’intervallo QT non è ancora del tutto chiarita.
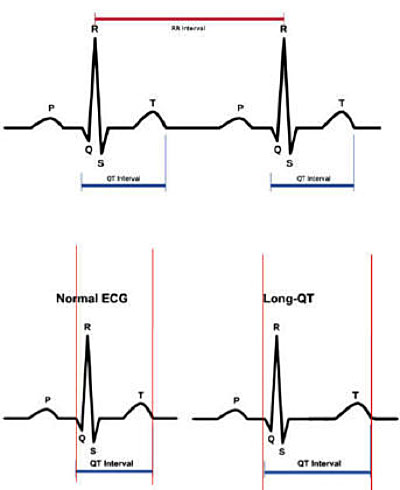
Per la correzione dell’intervallo QT per la frequenza cardiaca, oltre alla formula di Bazett, esiste anche la formula di Fredericia, particolarmente utilizzata nei pazienti pediatrici, e in particolare nell’analisi dell’intervallo QT in corso di registrazioni Holter, dal momento che la formula di Bazett può sovrastimare la durata dell’intervallo QT in caso di tachicardia marcata.
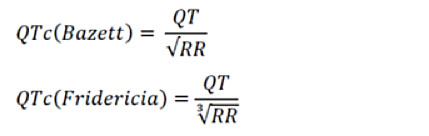
Nella LQTS, oltre che la durata dell’intervallo QT, è alterata anche la morfologia dell’onda T, con la comparsa di particolari anomalie, dette incisure o notches. La morfologia dell’ECG è stato descritto essere differente nelle diverse varianti genetiche (Moss, Circulation 1991).
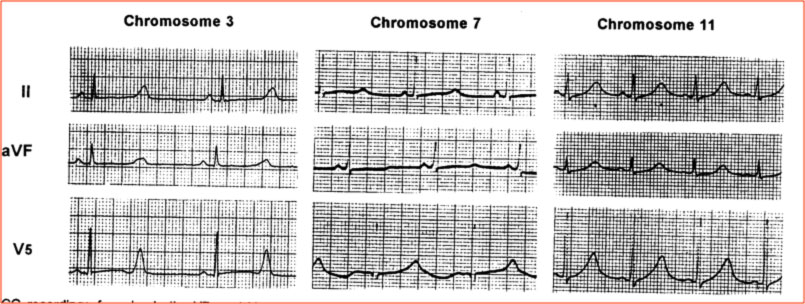
Differenti pattern ECG nei diversi genotipi della LQTS
Quali sono le caratteristiche genetiche della sindrome del QT lungo?
La Sindrome del QT lungo è causata da mutazioni su geni che controllano le correnti al potassio ed al sodio. Attualmente ne sono state identificate oltre 15 varianti genetiche, di cui i tre più importanti sono: KCNQ1 che determina la variante LQT1, KCNH2 (HERG) che determina la variante LQT2, e SCN5A che determina la variante più rara LQT3. Nelle forme familiari, varianti patogene di tali geni sono individuate in una percentuale di pazienti che va da 60% al 75% dei casi. Altre varianti genetiche sono indentificate in percentuali molto basse (circa 1-2% dei casi), mentre in circa 20% dei casi non si identificano mutazioni genetiche note.
| Genotipo | Gene | Cromosoma | Incidenza | Corrente |
| LQT1 | KCNQ1 | 11 | 30-35% | Iks (ridotta) |
| LQT2 | KCNH2 | 7 | 25-30% | Ikr (ridotta) |
| LQT3 | SCN5A | 3 | 5-10% | Ina (aumentata) |
L’analisi genetica è quindi fondamentale nella diagnosi della LQTS, ed è diventata una componente essenziale per il management clinico. L’identificazione del gene che provoca la malattia permette di effettuare uno screening familiare, per identificare rapidamente i familiari portatori della mutazione, permettendo in questo modo la prevenzione della morte improvvisa nell’intera famiglia.
Numerosi studi recenti indicano però che la LQTS non può essere sempre spiegata da una singola mutazione genetica, ma sembra seguire un modello genetico più complesso, dove polimorfismi genetici comuni potrebbero aver un effetto specifico sulla espressione della malattia (geni modificatori).
Molti studi infine sono già in atto per sviluppare trattamenti gene-specifici nella LQTS, mentre è già possibile una prevenzione gene-specifica del rischio di eventi, dato che i trigger degli eventi sono differenti nelle varie forme geniche.
Nel nostro Centro, l’analisi dei geni del QT lungo è effettuata mediante una piattaforma di “Next Generation Sequencing” (NGS) che permette di studiare contemporaneamente più pazienti per diversi geni. Tutte le varianti identificate mediante NGS sono sempre confermate con un’altra metodica di sequenziamento, il metodo Sanger. Se vuoi saperne di più sul nostro Servizio di Cardiogenetica clicca qui.
Quali sono i fattori scatenanti delle aritmie nella sindrome del QT lungo?
Vi è una stretta correlazione tra i fattori che scatenano le aritmie e la variante genetica di cui il paziente è portatore, pertanto la prevenzione del rischio di aritmie è diversa nelle differenti forme geniche.
Nei pazienti LQT1, il più numeroso sottogruppo genetico, la maggior parte degli eventi cardiaci potenzialmente letali avviene durante esercizio fisico, in particolare durante nuoto. I pazienti affetti da LQT1 sono inoltre sensibili alla deplezione di potassio.
I pazienti LQT2 sono particolarmente sensibili alle emozioni e ai rumori improvvisi, quali lo squillo del telefono o quello della sveglia e inoltre, le femmine LQT2 sembrano essere a più alto rischio nel periodo post-partum. I pazienti con LQT2 sono anche i più esposti al rischio di effetto proaritmico di alcuni farmaci, che agiscono come bloccanti della corrente Ikr, e della deplezione di potassio.
I pazienti LQT3 presentano più frequentemente eventi in condizioni di riposo o nel sonno. La terapia cardine è quella β-bloccante che ha dimostrato un’efficacia nella riduzione della mortalità nella popolazione di pazienti LQTS.
Quali sono i farmaci da evitare nella Sindrome del QT Lungo?
I pazienti con LQTS devono sempre evitare i farmaci che prolungano l’intervallo QT. Se vuoi conoscere la lista dei farmaci che sono da evitare nella sindrome del QT lungo clicca qui. Il sito che riporta tali informazioni aggiornate è il segunte: www.crediblemeds.org. Nei pazienti con LQTS è importante è la correzione dei disturbi elettrolitici (ipopotassiemia, ipomagnesiemia, ipocalcemia) che possono occorrere in caso di problemi malattie metaboliche o gastroenteriche, e in caso di terapie diuretiche.
Quali sono le terapie raccomandate nella Sindrome del QT Lungo?
La terapia farmacologica di prima scelta è basata sull’utilizzo dei farmaci beta-bloccanti a dose piena. Alcuni studi dimostrano che nella LQTS non tutti i β-bloccanti hanno pari efficacia e che i beta-bloccanti più efficaci sono propranololo e nadololo.
L’utilizzo di beta-bloccanti è raccomandati in tutti i soggetti con diagnosi clinica di LQTS, anche se asintomatici, dato che con i beta-bloccanti la mortalità, che nei sintomatici non trattati è di circa il 60%, diventa inferiore al 2%.
L’impianto di defibrillatore (ICD) è raccomandato nei pazienti con pregresso arresto cardiaco. L’impianto di ICD è anche raccomandato quando un paziente in trattamento con beta-bloccanti a dose piena ha una nuova sincope. L’ICD è infine consigliato nei pazienti asintomatici con QT molto prolungato (QTc > 500 msec), portatori di una mutazione patogena dei tre geni principali.
Nei pazienti portatori di ICD che abbiano una recidiva aritmica, o in pazienti con controindicazioni a beta-bloccanti o ICD, può essere considerata l’esecuzione della denervazione cardiaca simpatica di sinistra.
Bloccanti dei canali del sodio (Mexiletina o Flecainide o Ranolazina) possono essere considerati come terapia additiva per accorciare l’intervallo QT in pazienti LQT3 con QTc maggiore di 500 msec.
Bibliografia
- Moss AJ, Schwartz PJ, Crampton RS, Tzivoni D, Locati EH, MacCluer J, Hall WJ, Weitkamp L, Vincent GM, Garson A, Jr. and et al. The long QT syndrome. Prospective longitudinal study of 328 families. Circulation 1991; 84:1136-1144.
- Priori, SG, Wilde AA, Horie M et al: HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndrome. Heart rhythm 2013; 10: 287-294.
- Moss AJ, Zareba W, Benhorin J, Locati EH, Hall WJ, Robinson JL, Schwartz PJ, Towbin JA, Vincent GM, Lehmann M, Keating MT, MacCluer JW, Timothy KW: ECG T wave patterns in genetically distinct forms of the hereditary long QT syndrome. Circulation 1995; 92: 2929-2934.
- Schwartz PJ, Priori SG, Locati EH, Napolitano C, Cantu’ F, Towbin AJ, Keating MT, Hammoude H, Brown AM, Chen LK, Cotasky TJ: Long QT syndrome patients with mutations on the SCN5A and HERG genes have differential responses to Na+ channel blockade and to increase in heart rate. Implications for gene-specific therapy. Circulation 1995; 92: 3381-86.
- Mizusawa Y, Horie M, Wilde AAM, Genetic and Clinical Advances in Congenital Long QT Syndrome., Circ J 2014; 78: 2827–2833.