Cardiomiopatia o Displasia Aritmogena (DAVD o ARVD)
Indice dell'articolo
Che cos’è la cardiomiopatia (o displasia) aritmogena del ventricolo destro (ARVD)
La cardiomiopatia (detta anche displasia) aritmogena del ventricolo destro (ARVD) è una cardiopatia del muscolo cardiaco, caratterizzata clinicamente da aritmie ventricolari potenzialmente letali. La prevalenza è stimata di 1:2.000, anche se in alcuni paesi (Italia e Grecia) la malattia è particolarmente comune (anche 1:700). La malattia consiste in una degenerazione del miocardio ventricolare, prevalentemente localizzata al ventricolo destro (VDx), ma che può coinvolgere possibile anche al ventricolo sinistro (VSx). Il tessuto muscolare cardiaco (detto miocardio) viene sostituito da un tessuto fibro-adiposo di tale entità da causare aneurismi (dilatazioni) del ventricolo. Non è agevole stimare la reale prevalenza ed incidenza della ARVD perché i pazienti non sempre sono facilmente inquadrabili dal punto di vista diagnostico. Inoltre talvolta la prima manifestazione della patologia è proprio la morte cardiaca improvvisa e questo complica le indagini epidemiologiche. La prevalenza della malattia è simile nei due sessi, anche se la maggioranza dei pazienti sintomatici è di sesso maschile.
Qual’è la presentazione clinica della ARVD?
La ARVD è una delle cause maggiori di morte improvvisa nei giovani adulti e negli atleti. La sua presentazione clinica solitamente consiste in fenomeni aritmici che variano dalla extrasistolia ventricolare isolata più o meno frequente alla tachicardia o fibrillazione ventricolare (TV/FV). Lo spettro di presentazione è comunque molto variabile, ed include oltre alle aritmie e alle alterazioni globali o segmentali del VDx, anche tipiche alterazioni elettrocardiografiche della depolarizzazione e ripolarizzazione (visibili soprattutto nelle derivazioni precordiali, e una evoluzione in scompenso cardiaco, che può mimare una cardiomiopatia dilatativa di altra origine.
Quali sono le caratteristiche genetiche della ARVD?
In base alle attuali conoscenze, la ARVD ha una base genetica, e non sono note forme acquisite. Sono stati descritti numerosi geni che, quando mutati, provocano la ARVD. Tra questi rientrano i geni PKP2, DSG2, DSC2, TGFB3, DSP, JUP, TMEM43; più recentemente si sono aggiunti anche i geni LDB3, LMNA, RYR2, TTN e CTNNA3. I geni che causano la malattia codificano per le proteine delle giunzioni cellulari meccaniche (placoglobina, placofilina, desmogleina, desmocolina, desmoplakina).
Una storia familiare di ARVD è presente in una percentuale che va dal 30 al 50% dei casi. Il più comune pattern di trasmissione è autosomico dominante con penetranza variabile ed espressione fenotipica polimorfica, sebbene sia stato anche descritto un modello autosomico recessivo. La più comune modalità ereditaria della ARVD è di tipo autosomico dominante: è sufficiente possedere mutata una singola copia di un gene-malattia (mutazione in eterozigosi) per risultare affetti. In molte famiglie con ARVD a genotipo noto sono state riscontrate penetranza incompleta (non tutti gli eterozigoti per mutazioni causanti presentano manifestazioni cliniche) ed un’espressività variabile (il quadro clinico può variare anche fra individui correlati geneticamente ed eterozigoti per la medesima mutazione causante). Oltre alla tipica forma con trasmissione autosomica dominante e penetranza variabile, sono state anche osservate forme recessive associate a cheratoderma palmo-plantare e capelli lanosi.
Come viene effettuata la diagnosi di ARVD?
La diagnosi di ARVD solitamente avviene in età giovanile o adulta; A causa dell’evoluzione progressiva della patologia, i pazienti spesso presentano un corredo sintomatologico eterogeneo. Palpitazioni, fatica e sincope, sembrano essere i sintomi più comuni, ma talora sono presenti disturbi aspecifici come dolore addominale e confusione mentale. In alcuni casi però, la prima manifestazione può essere un arresto cardiaco in concomitanza con sforzi fisici intensi, oppure nelle ore notturne. Le principali diagnosi differenziali si pongono con le miocarditi, la cardiomiopatia dilatativa, la sarcoidosi e l’amiloidosi.
La diagnosi dell’ARVD comincia con una buona annotazione anamnestica. Una storia personale di palpitazioni (specie in persone di giovane età), con familiarità per morte improvvisa dovrebbe sempre indurre al sospetto della patologia in questione.
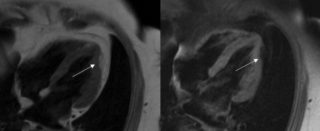
Gli strumenti di elezione per la diagnosi sono i test di imaging, inclusa l’ecografia bidimensionale, l’angiografia e la risonanza magnetica, che rivelano le anomalie strutturali e funzionali. La mappa elettroanatomica permette di individuare le aree a basso voltaggio corrispondenti a un’atrofia del miocardio, con sostituzione grasso-fibrosa.
Quali sono le anomalie elettrocardiografiche tipiche della ARVD?
Dal 50 al 90% dei pazienti presentano anomalie elettrocardiografiche, che includono:
1) inversione dell’onda T nelle derivazioni precordiali, in soggetti di età > 12 anni ed in assenza di BBDx
2) onda epsilon
3) extrasistoli ventricolari frequenti (>1000 BEV/24 ore) con morfologia a blocco di branca sinistro,
4) tachicardie ventricolari non sostenute (TVNS) o tachicardie ventricolari sostenute (TVS) con morfologia a blocco di branca sinistro (anche se è possibile riscontrare TV con morfologie differenti).
5) Potenziali tardivi all’ECG ad alta risoluzione
Quali sono i fattori di rischio per la prognosi della ARVD?
I maggiori fattori di rischio per una prognosi infausta sono la giovane età, i precedenti familiari di morte improvvisa giovanile, un QRS maggiore o uguale a 140 ms, l’inversione dell’onda T sul tracciato, l’entità del coinvolgimento del ventricolo destro, il coinvolgimento del ventricolo sinistro, la presenza di tachicardie ventricolari, la storia di sincope o di un precedente arresto cardiaco.
Lo studio elettrofisiologico endocavitario (SEE) è utilizzato per una corretta stratificazione del rischio al fine di valutare la suscettibilità del substrato aritmogeno. Con lo SEE è possibile determinare il tipo di aritmia inducibile, la sua morfologia e la tollerabilità emodinamica.
Qual è l’evoluzione clinica della ARVD?
La storia naturale dell’ARVD dipende sia dall’instabilità elettrica del substrato sia dalla progressiva disfunzione ventricolare. Con l’evoluzione della patologia si assiste ad una alterazione della contrattilità ventricolare che residua in una insufficienza cardiaca destra o biventricolare. L’interessamento del ventricolo sinistro infatti, sia esso macroscopico o istologico, interessa circa il 70% dei pazienti affetti. Questo coinvolgimento sembra essere età dipendente, e l’esercizio fisico intenso (ad esempio negli atleti agonisti) sembra peggiorare e accelerare l’evoluzione clinica della malattia. E’ quindi raccomandato seguire l’evoluzione clinica con indagini ecocardiografiche seriate. Nella storia naturale comunque potrebbero essere considerate le seguenti fasi:
1) fase inziale o occulta, caratterizzata da alterazioni strutturali minime, con o senza aritmie ventricolari minori, durante la quale la morte improvvisa potrebbe essere occasionalmente la prima manifestazione di malattia (in particolare durante attività fisica intensa);
2) fase aritmogena, durante la quale le aritmie ventricolari ad origine dal ventricolo destro sono sintomatiche e possono portare ad arresto cardiaco (in presenza o meno di anomalie funzionali del ventricolo);
3) fase di disfunzione ventricolare destra, con la progressiva comparsa di insufficienza del ventricolo destro dovuta alla progressiva sostituzione del muscolo con tessuto fibroadiposo, con una funzione ventricolare sinistra relativamente conservata;
4) fase di disfunzione biventricolare, in cui si assiste ad un progressivo coinvolgimento e disfunzione biventricolare. In questa fase l’ARVD può mimare una diversa cardiomiopatia dilatativa e portare a scompenso cardiaco congestizio con tutte le sue relative complicanze quali fibrillazione atriale ed eventi tromboembolici.
Quali sono le terapie della ARVD?
Il principale trattamento della displasia aritmogena è rivolto alla prevenzione della morte cardiaca improvvisa. Sebbene non ci sia modo di curare l’ARVD, è possibile controllare le manifestazioni aritmiche e la disfunzione ventricolare. Le terapie disponibili includono modificazioni dello stile di vita, farmaci antiaritmici, ablazione del substrato aritmico con radiofrequenza, impianto di un defibrillatore automatico (ICD).
Sebbene non esistano evidenze definitive riguardo le modificazioni dello stile di vita, i pazienti dovrebbero evitare attività fisica intensa, sia per evitare aritmie, che per evitare il sovraccarico ventricolare che può favorire la disfunzione ventricolare.
Nel caso di aritmie possono essere utilizzati i farmaci antiaritmici, che peraltro hanno mostrato un’efficacia limitata. L’amiodarone somministrato per via endovenosa risulta efficace nell’interrompere le tachicardie ventricolari. Altri regimi farmacologici comprendono farmaci betabloccanti, da soli o in associazione con farmaci di classe Ia e Ic, amiodarone in associazione a farmaci di classe II o Ic.
L’ablazione mediante radiofrequenza è utilizzata in caso di tachicardie ventricolari incessanti o refrattarie al trattamento medico, tachicardie ventricolari ben localizzabili, successivamente all’impianto del defibrillatore per ridurre gli interventi dello stesso. L’efficacia dell’ablazione varia da caso a caso ed a volte è necessaria più di una procedura. Le recidive spesso sono causa dell’evoluzione stessa della patologia che crea nuovi circuiti di rientro.
L’impianto di un defibrillatore in generale è raccomandato nei pazienti, oltre che in prevenzione secondaria in caso di pregresso arresto cardiaco resuscitato, in prevenzione primaria nei soggettivi che hanno inducibilità ad aritmie ventricolari sostenute durante SEE, nei soggetti con aritmie ventricolari non sostenute e storia familiare di morte improvvisa di giovane età e qualora ci sia coinvolgimento del ventricolo sinistro.