La cardiomiopatia ipertrofica (CMI) è una malattia del muscolo cardiaco caratterizzata da un aumento dello spessore delle pareti del ventricolo sinistro (ipertrofia ventricolare sinistra, spesso a livello del setto interventricolare o dell’apice cardiaco), non secondario a ipertensione arteriosa, a malattie delle valvole cardiache o ad altre cause sistemiche che possano generare ipertrofia ventricolare sinistra. I pazienti presentano un esordio subdolo con aritmie e la morte improvvisa può costituire il primo segno clinico della condizione. L’incidenza è difficile da calcolare perché molti pazienti presentano un quadro sindromico, con anomalie a carico di altri organi ed apparati.
Lo sviluppo dell’ipertrofia avviene solitamente nell’adolescenza o in età adulta, nelle femmine più tardivamente, fino alla sesta o settima decade, mentre solo una minoranza dei pazienti presenta ipertrofia ipertrofia già alla nascita o in età pediatrica. La diagnosi di cardiomiopatia ipertrofica viene posta mediante esame ecocardiografico o risonanza magnetica, quando si osserva uno spessore di parete del ventricolo sinistro superiore a 15 mm in uno o più segmenti del ventricolo sinistro (con maggiore frequenza nel setto interventricolare, più raramente a livello apicale o diffusamente in tutte le pareti).
La malattia può esordire ad ogni età con una rilevante percentuale di pazienti asintomatici che hanno la morte improvvisa come unica manifestazione della patologia. Nei pazienti sintomatici essa può presentarsi con dispnea, cardiopalmo, sincope e dolore precordiale. Alterazioni aspecifiche del tracciato ECG (con alti voltaggi del QRS e onde T negative) e aritmie (tachicardia da rientro nodale, fibrillazione e flutter atriale, aritmie ventricolari) possono presentarsi in molti pazienti, specialmente quelli che possiedono mutazioni in singola copia nel gene MYBPC3.
QUAL’E’ LA DIFFUSIONE DELLA CARDIOMIOPATIA IPERTROFICA?
La CMI è una forma clinica nota da secoli ed ancor oggi è una delle principali cause di morte improvvisa. La CMI è una delle più comuni forme di malattia genetica cardiaca. La prevalenza della cardiomiopatia ipertrofica nella popolazione adulta, valutata in diverse aree geografiche e differenti popolazioni (dagli Stati Uniti al Giappone alla Tanzania), è di circa il 2 per mille: quindi, la cardiomiopatia ipertrofica non è considerata una malattia rara.
QUALI SONO LE BASI GENETICHE DELLA CARDIOMIOPATIA IPERTROFICA?
Le cause di CMI sono quasi soltanto genetiche e tra queste possono essere sindromiche o non sindromiche. Tra le forme sindromiche abbiamo la malattia di Fabry, la sindrome di Noonan e la sindrome Leopard. Una delle sindromi più comunemente associate a cardiomiopatia ipertrofica è la sindrome di Noonan (in media 1/1700 nati vivi). Tra le forme non sindromiche possiamo trovare l’amiloidosi e le forme isolate di ipertrofia settale (tra le più comuni in Europa ed Usa).
La CMI è causata da mutazioni in singola o duplice copia dei geni che codificano le proteine muscolari cardiache. I geni più comunemente implicati sono MYBPC3, MYH7 e TNNT2, responsabili da soli di almeno il 60% dei casi. Un paziente affetto dalla CMI in genere possiede mutata una sola copia del gene causativo. Ne consegue che il rischio di ricorrenza è generalmente pari al 50% per ogni gravidanza. il gene MYBPC3: quest’ultimo è stato ritrovato mutato in una decina di nostri pazienti affetti dalla sindrome di Brugada, suggerendo un substrato genico condiviso tra le due patologie (unpublished data).
La modalità di trasmissione della CMI è di solito “autosomica dominante”. Questo significa che nella maggioranza dei casi la malattia si trasmette da genitore a figlio/a con una probabilità del 50%. Tuttavia, in alcune famiglie è possibile che il genitore o il figlio non presentino un’espressione clinica evidente della malattia. Questo fenomeno è definito “penetranza incompleta” e significa che non sempre la patologia si manifesta in tutti i pazienti portatori del difetto genetico.
QUALI SONO I CRITERI DIAGNOSTICI DELLA CARDIOMIOPATIA IPERTROFICA?
La diagnosi clinica si fonda sul riconoscimento di un ipertrofia ventricolare (in presenza o in assenza di storia familiare positiva). Essa viene definita come spessore della parete ventricolare sinistra pari o maggiore di 15 mm nei pazienti adulti. Nel paziente pediatrico si utilizza uno score che calcola le deviazioni standard per età. In tali pazienti lo spessore del setto è patologico qualora eguale o maggiore di 2 deviazioni standard per età. È fondamentale escludere l’ipertensione arteriosa e la stenosi della valvola aortica in un paziente di qualsiasi età. Nei casi con storia familiare i criteri sono meno stringenti consentendo una diagnosi clinica con uno spessore ventricolare pari o superiore a 13 mm. È importante sapere che alcune anomalie valvolari (per esempio prolasso mitralico o cleft miocardico) possono supportare un sospetto diagnostico anziché escluderlo.
QUALI SONO I SINTOMI DELLA CARDIOMIOPATIA IPERTROFICA?
Inizialmente, la cardiomiopatia ipertrofica è spesso asintomatica, oppure i sintomi possono essere mal interpretati dal paziente. In questi casi, la diagnosi viene posta durante controlli eseguiti per altri motivi, come ad esempio in occasione di una visita medico sportiva. I sintomi più comuni, quando presenti, sono: palpitazioni, dispnea (“mancanza di fiato”) durante lo sforzo, dolore toracico a riposo o da sforzo, sincope (“svenimento improvviso”), spesso durante lo sforzo
Nella maggior parte dei casi la cardiomiopatia ipertrofica è una cardiopatia relativamente benigna, con una stabilità di sintomi e della stessa ipertrofia nelle decadi successive alla diagnosi. Tuttavia, nel corso degli anni circa il 20% dei pazienti può presentare un peggioramento dei sintomi e complicanze come aritmie, sia atriali (in particolare la fibrillazione atriale) che ventricolari sia extrasistoli ventricolari, che tachicardie ventricolari non sostenute o sostenute, che possono causare sincope (perdita di coscienza), che fibrillazione ventricolare (che provoca arresto cardiaco e morte improvvisa).
QUAL’E’ ITER DIAGNOSTICO DELLA CARDIOMIOPATIA IPERTROFICA?
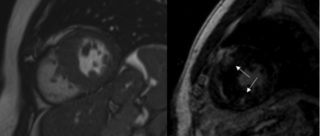
Cardiomiopatia ipertrofica con evidenza di aree di late gadolinium enhancement in corrispondenza delle giunzioni con la parete libera ventricolare destra indicative di sostituzione fibrotica.
Questi accertamenti consentono di valutare la severità della cardiopatia e il rischio potenziale di complicanze. A questi dati si aggiungono le informazioni fornite dall’esame genetico (effettuato con il consenso del paziente dopo laconsulenza genetica).
Una volta posta la diagnosi, i parenti di I grado del paziente (genitori, fratelli o sorelle, figli) vengono invitati a eseguire gli esami cardiologici di base (visita, elettrocardiogramma ed ecocardiogramma) in modo da valutare se anche alcuni tra loro presentino segni di cardiomiopatia ipertrofica. Quando la mutazione genetica responsabile è stata identificata nel paziente viene ricercata anche nei familiari di I grado. Il contributo dei test genetici è importante nello screening familiare perché permette di identificare i familiari portatori della mutazione genetica responsabile della malattia prima che compaiano sintomi.
QUALI SONO LE TERAPIE DELLA CARDIOMIOPATIA IPERTROFICA?
Al momento non esiste una cura che guarisca la cardiomiopatia ipertrofica, tuttavia esistono terapie per migliorare i sintomi e prevenire le eventuali complicanze. La necessità e la scelta del trattamento è personalizzata, differente per ciascun paziente. I farmaci più utilizzati nei pazienti sono i beta-bloccanti e i calcio-antagonisti. In caso di fibrillazione atriale possono essere necessari farmaci anticoagulanti. In caso di aritmie possono essere utilizzati farmaci antiaritmici di classe 1a e 1c, più raramente l’amiodarone. In caso di scompenso cardiaco possono essere utilizzati diuretici e ACE-inibitori.
QUANDO È NECESSARIO L’IMPIANTO DI DEFIBRILLATORE NELLA CARDIOMIOPATIA IPERTROFICA?
I pazienti con CMI ad alto rischio di aritmie ventricolari e morte improvvisa sono candidati all’impianto di un defibrillatore automatico (Implantable Cardioverter-Defibrillator o ICD). Sono candidati all’impianto di ICD sia pazienti sopravvissuti ad un arresto cardiaco (prevenzione secondaria) sia pazienti in prevenzione primaria considerati ad alto rischio per la presenza di aritmie ventricolari ripetitive o con storia familiare di morte improvvisa dovuta alla cardiomiopatia ipertrofica. L’ICD è in grado di identificare aritmie potenzialmente letali e dare uno shock elettrico (o di fare una stimolazione antitachicardica) per interrompere queste aritmie e reinstaurare il normale ritmo cardiaco. I pazienti devono comunque essere informati riguardo alle possibili complicanze associate all’ICD, che includono scariche elettriche inappropriate durante frequenze cardiache elevate ma normali (attualmente, questi interventi inappropriati avvengono in quasi il 25% dei pazienti), spesso dovuti a problemi con i cateteri (usualmente, rottura del catetere, relativamente frequenti in questi pazienti). Il periodo di esposizione al rischio di morte improvvisa è tipicamente molto lungo nella CMI (teoricamente 20-50 anni in alcuni pazienti), ed è quindi probabile che l’ICD intervenga per la prima volta solo dopo anni (5-10 anni) dal momento dell’impianto.
QUANDO È NECESSARIA LA CHIRURGIA NELLA CARDIOMIOPATIA IPERTROFICA?
Circa il 30% dei pazienti con cardiomiopatia ipertrofica presenta già a riposo una ostruzione al tratto d’efflusso ventricolare sinistro che può essere valutata e misurata con l’ecografia transtoracica. L’ipertrofia del setto interventricolare, unita allo spostamento dell’apparato valvolare mitralico, riduce la dimensione della zona di passaggio tra ventricolo sinistro ed aorta (“tratto di efflusso”), così da rendere necessaria una pressione più elevata (“gradiente”) per espellere il sangue dal ventricolo verso l’aorta.
L’ostruzione può essere assente a riposo ma presentarsi durante sforzo in un ulteriore 30% dei pazienti, e questo fenomeno può essere valutato con l’esame ecocardiografico ColorDoppler eseguito durante test da sforzo.
Se l’ostruzione è severa e causa sintomi importanti (capogiro, svenimenti, dolore toracico), può essere indicata la sua riduzione, che si effettua solitamente con un intervento cardiochirurgico (miectomia), da eseguirsi in Centri con consolidata esperienza.
“Trattiamo le aritmie cardiache dallo studio dei geni all’ablazione transcatetere“