La sindrome di Noonan

La sindrome di Noonan è una malattia genetica pleomorfa a trasmissione autosomica dominante caratterizzata da una facies caratteristica, bassa statura, difetti cardiaci congeniti e ritardo dello sviluppo di grado variabile.
Nel 1968, la dott.ssa Jacqueline Noonan descrisse la sindrome omonima, raggruppando nove pazienti che mostravano una serie di caratteristiche cliniche simili associate a cardiopatie congenite più o meno complesse.
Queste caratteristiche, che possono essere più o meno pronunciate , includono una facies tipica ( ipertelorismo, ptosi , orecchie basse, forma triangolare del viso), bassa statura e anomalie del torace (pectus excavatum inferiore o pectus carinatum superiore).
Ulteriori caratteristiche extracardiache includono disabilità dello sviluppo neurologico, criptorchidismo, pubertà ritardata, linfedema, disturbi emorragici e una predisposizione leggermente aumentata a tumori ematologici e solidi (Roberts, Allanson, Tartaglia e Gelb, 2013).
È stato stabilito che la trasmissione sia autosomica dominante, sebbene sia stata recentemente identificata una forma autosomica recessiva (Johnston et al., 2018). Il fenotipo è variabile, ma si ritiene che la penetranza sia completa.
La sindrome di Noonan condivide molti aspetti con altre sindromi quali la sindrome di Costello, sindrome cardiofaciocutanea, la sindrome di Noonan con lentiggini multiple (precedentemente nota come sindrome LEOPARD).
Tali patologie sono contraddistinte da una medesima fisiopatologia secondaria a mutazione dei geni nella via del signaling RAS/MAPK. Nell’insieme esse costituiscono le così dette Rasopatie.
Dal 2001, quando è stato scoperto il PTPN11, il primo gene identificato per la Sindrome di Noonan (Tartaglia et al., 2001), il grado di eterogeneità molecolare alla base delle RASopatie è diventato evidente.
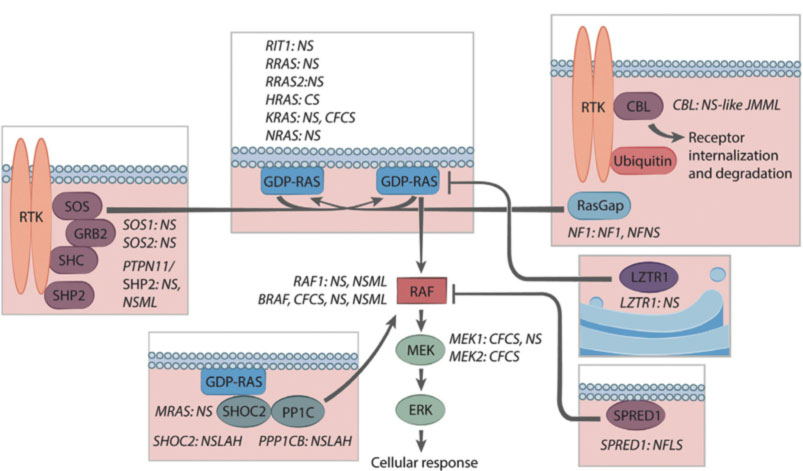

Una delle principali caratteristiche della NS, identificata fin dalla prima descrizione, è il coinvolgimento cardiaco.
I pazienti affetti presentano un ampio spettro di malattie cardiache, che rientrano in due categorie principali: cardiopatie congenite (CHD), presente in circa l’80% dei pazienti, e cardiomiopatia ipertrofica (HCM), riscontrata in circa il 20% dei pazienti (Marino et al., 1999).
La cardiopatia congenita si manifesta molto spesso come stenosi della valvola polmonare (PVS), con una prevalenza stimata di ~ 40%, ma sono stati descritti altri tipi, in particolare difetti del setto interatriale (8%) o interventricolare (ASD / VSD), nonché difetti del canale atrioventricolare (AVCD) (15%) (Calcagni et al., 2017).
Meno frequentemente, i pazienti presentano forme di cardiopatia che interessano le strutture cardiache sinistre tra cui la stenosi della valvola mitrale (6%), stenosi della valvola aortica (AS) e coartazione aortica (9%) (Lam, Corno, Oorthuys e Marcelletti, 1982).
Dopo la delucidazione delle varianti genetiche responsabili della SN, è diventato possibile cercare diverse associazioni genotipo-fenotipo. Ne sono emerse in discreto numero anche se alcune associazioni sono lontane dall’esser chiaramente identificate a causa dell’esiguità del numero dei pazienti.
Le varianti patogene del PTPN11, ad esempio, costituiscono quasi l’80% delle cause identificate di NS tra i pazienti che presentano stenosi della valvola polmonare o difetti interatriali (Prendiville et al., 2014); al contrario, le mutazioni a carico di PTPN11 sono negativamente associate con la cardiopatia ipertrofica HCM (6% vs 26% dei pazienti afetti non PTPN11) (Tartaglia et al., 2002).
Le varianti patogene del gene RAF1 sono fortemente associate alla miocardiopatia ipertrofica ma negativamente associate a PVS (Pandit et al., 2007).
La variazione patogena in RIT1 predispone i pazienti ad anomalie valvolari che alla cardiopatia ipertrofica
La patogenesi delle CHD nella sindrome di Noonan , in particolare per quanto riguarda l’eterogeneità dei difetti cardiaci è solo parzialmente compresa. Per il prodotto del gene PTPN11, la proteina tirosina fosfatasi SHP2, è stato stabilito un chiaro ruolo nella morfogenesi valvolare.
In particolare, l’attivazione della via MAPK attraverso i recettori del fattore di crescita epidermico costituisce il meccanismo principale che guida la transizione epitelio-mesenchimale (EMT) nella formazione della valvola semilunare (Krenz, Yutzey e Robbins, 2005).
Clinical Case
Nel novembre del 2020 giungeva presso il pronto soccorso del nostro istituto un ragazzo di 20 anni per un episodio sincopale intervenuto a riposo.
La TAC encefalo e l’esame neurologico non mostravano alcuna acuzie e pertanto veniva indirizzato alla nostra attenzione.
L’ECG ottenuto all’ingresso mostrava chiari segni di ipertrofia ventricolare sinistra. Per tale motivo veniva richiesto anche un approfondimento mediante ecocardiografia.
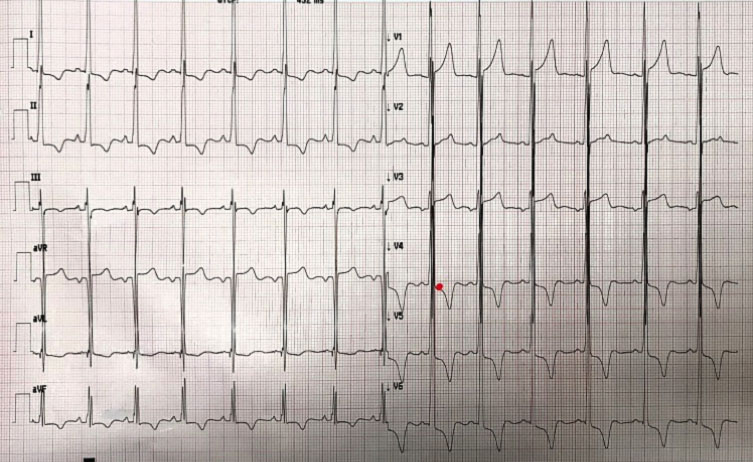
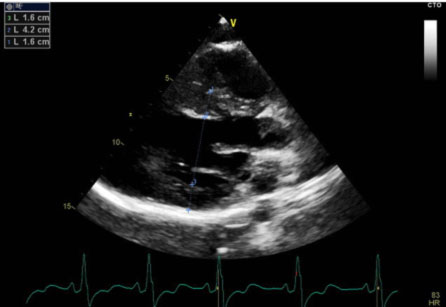
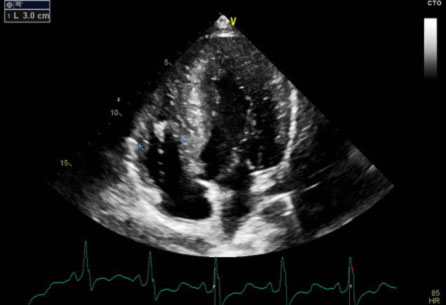
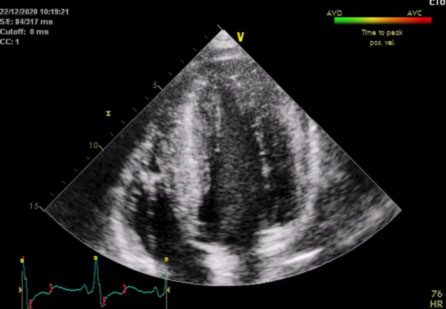
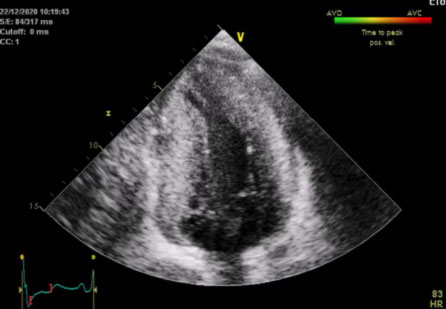
L’ecocardiografia mostrava i seguenti parametri: Severa ipertrofia concentrica del ventricolo sinistro con trabecolatura dei segmenti medio apicali, con ipertrofia a distribuzione asimmetrica prevalentemente localizzata a livello del setto interventricolare inferiore (21 mm) e della parete inferiore. La parete posteriore mostra spessore di ca. 16 mm. Funzione sistolica del ventricolo sinistro ai limiti inferiori di norma (FE 50%). Non segni di ostruzione all’efflusso sinistro. Pattern diastolico da alterato rilasciamento con normali pressioni di riempimento (E/e’ < 10). Atrio sinistro non dilatato. Radice aortica ed aorta ascendente di normali dimensioni. Ventricolo destro non dilatato, con lieve deficit della funzione sistolica (TAPSE 16 mm). Vena cava inferiore non dilatata e con normali escursioni respiratorie.
Il quadro ecocardiografico era pertanto compatibile con la diagnosi di miocardiopatia ipertrofica non ostruttiva.
 |
 |
 |
 |
Tale riscontro rendeva suggestiva l’ipotesi di una sincope a genesi aritmica. Per tale motivo si disponeva ricovero al fine di eseguire ulteriori accertamenti diagnostici.
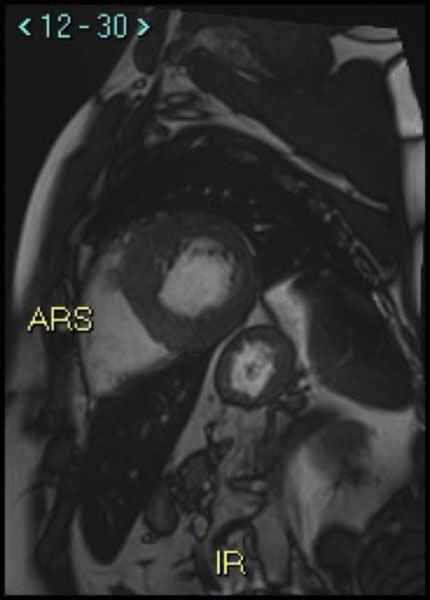
La risonanza magnetica cardiaca (CMRI) con mezzo di contrasto mostrava quadro di miocardiopatia ipertrofica con estesa fibrosi miocardica localizzata in prossimità delle parete inferosettali, anteriore ed anterolaterale (LGE> 30% della massa miocardica totale).
 |
 |
La presenza di una così vasta area di fibrosi miocardica pone il paziente ad un aumentato rischio di morte cardiaca improvvisa secondaria ad aritmie ventricolari sostenute (fibrillazione ventricolare o tachicardia ventricolare).
In considerazione dell’episodio sincopale si decideva pertanto di impiantare un defibrillatore in prevenzione primaria.
Ad un’esame fisico attento inoltre il paziente mostrava alcune caratteristiche peculiari : Bassa statura , fronte ampia, solchi naso genieni molto evidenti.
La facies complessiva suggeriva un’aspetto sindromico compatibile con la sindrome di Noonan.
Richiesta quindi la consulenza del collega genetista si procedeva anche ad espletare i relativi test genetici.
I test genetici mostravano una mutazione in eterozigosi a carico del gene SOS1 (Ras/Rac guanine nucleotide exchange factor 1).